Lofexidine (Lofetensin; Loxacor; brand name Lucemyra among others), an analog of Clonidine, is a medication historically used to treat high blood pressure, but nowadays, it is more commonly used to help with the physical symptoms of opioid withdrawal. Lofexidine is an agonist of the α2A adrenergic receptor that the Food and Drug Administration of the United States approved for use in 2018. Although it has been studied, the efficacy of lofexidine as a treatment for alcohol withdrawal symptoms has not yet been demonstrated. Additionally, postmenopausal hot flash sufferers are treated with it.
Physicochemical Properties
| Molecular Formula | C11H12CL2N2O |
| Molecular Weight | 259.13 |
| Exact Mass | 258.032 |
| Elemental Analysis | C, 50.99; H, 4.67; Cl, 27.36; N, 10.81; O, 6.17 |
| CAS # | 31036-80-3 |
| Related CAS # | Lofexidine hydrochloride; 21498-08-8 |
| PubChem CID | 30668 |
| Appearance | White to off-white solid powder |
| Density | 1.4±0.1 g/cm3 |
| Boiling Point | 421.5±35.0 °C at 760 mmHg |
| Melting Point | 215-225 °C |
| Flash Point | 208.7±25.9 °C |
| Vapour Pressure | 0.0±1.0 mmHg at 25°C |
| Index of Refraction | 1.611 |
| LogP | 3.59 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 2 |
| Rotatable Bond Count | 3 |
| Heavy Atom Count | 16 |
| Complexity | 263 |
| Defined Atom Stereocenter Count | 0 |
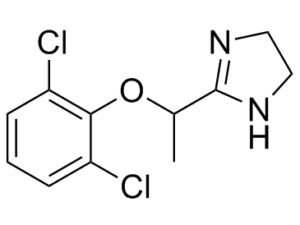
| SMILES | CC(C1=NCCN1)OC2=C(Cl)C=CC=C2Cl |
| InChi Key | KSMAGQUYOIHWFS-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C11H12Cl2N2O/c1-7(11-14-5-6-15-11)16-10-8(12)3-2-4-9(10)13/h2-4,7H,5-6H2,1H3,(H,14,15) |
| Chemical Name | 2-[1-(2,6-dichlorophenoxy)ethyl]-4,5-dihydro-1H-imidazole |
| Synonyms | RMI-14042A; MDL-14,042; MDL 14,042; 31036-80-3; Lofexidina; Lofexidinum; 2-[1-(2,6-dichlorophenoxy)ethyl]-4,5-dihydro-1H-imidazole; Lofexidinum [INN-Latin]; Lofexidina [INN-Spanish]; 2-(1-(2,6-Dichlorophenoxy)ethyl)-4,5-dihydro-1H-imidazole; MDL14,042; Lofexidine; Loxacor; RMI14042A; RMI 14042A; BA 168; Lofetensin; Lucemyra |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | α2-receptor |
| ln Vitro | Lofexidine is one therapeutic option used for treating the onslaught of sympathetic outflow that typically commences upon induction of opiate withdrawal. It was approved for opiate detoxification in the UK, most of EU, and a select few countries worldwide during the 1980s and the 90s. Within the US and Canada, however, it remains an experimental drug[1]. |
| ln Vivo | Lofexidine is an alpha(2)-agonist structurally related to clonidine. It is not an effective antihypertensive agent; however, it decreases the sympathetic outflow responsible for many opioid withdrawal symptoms. Nine clinical studies were reviewed representing 354 patients receiving lofexidine including a recent Phase 3 clinical trial. Eight studies involved comparisons of lofexidine to an opioid receptor agonist or clonidine for opioid detoxification. In these trials, lofexidine dosing was titrated to a maximum of 1.6-3.2 mg/day in divided doses for a total of 5-18 days. The data suggest that lofexidine has positive efficacy in reducing opioid withdrawal symptoms and is at least as effective as the opioid receptor agonists utilized for detoxification. Not all withdrawal symptoms are alleviated by alpha(2)-agonists, with many patients complaining of insomnia and aching. The most common adverse event with lofexidine in the Phase 3 trial was insomnia. Hypotension was also reported; however, the studies comparing clonidine with lofexidine suggest decreased incidence and severity of adverse events with lofexidine[2]. |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Lofexidine has a good oral bioavailability and the peak plasma concentration occurs after 2-5 hours of oral administration. The bioavailability is registered to be even higher than 72%. About 30% of the administered dose of lofexidine is lost during first-pass metabolism. The absorption is registered to be very rapidly recirculated in the gut. After oral administration of 0.8 mg of lofexidine, a maximal dose of 1.26 ng/ml is achieved after 3 hours. The elimination of lofexidine is primarily through the renal system and it represents 94% of the administered dose while elimination in feces corresponds to only 0.93%. From the eliminated dose in urine, about 10% is formed by unchanged drug and 5% is constituted by the first hydrolysis product N-(2-aminoethyl)-2-(2,6-dichlorophenoxy)propanamide. 2,6-dichlorophenol represents the majority of the administered dose by occupying about 80% of the administered dose. Lofexidine has a volume of distribution of 300 L, indicating that it distributes readily into the tissues. The total elimination clearance following intravenous administration is 17.6 L/h. Metabolism / Metabolites Lofexidine metabolic ratio is highly variable among people. It is metabolized mainly by the activity of CYP2D6 and in a minor degree by CYP1A2 and CYP2C19. These enzymes catalyze the hydroxylation of lofexidine and the opening of imidazoline ring to form N-(2-aminoethyl)-2-(2,6-dichlorophenoxy)propanamide. This metabolite is deamidated and forms 2-(2,6-dichlorophenoxy) propionic acid and 2,6-dichlorophenol. These three main metabolites are inactive. Biological Half-Life The reported elimination half-life of lofexidine is 11 hours. |
| Toxicity/Toxicokinetics |
Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the use of lofexidine during breastfeeding. Lofexidine is chemically and pharmacologically related to clonidine, which reaches high levels in milk and in the breastfed infant. Although it is not contraindicated during breastfeeding, lofexidine should be used with caution during breastfeeding, especially while nursing a newborn or preterm infant. An alternate drug may be preferred, especially while nursing a newborn or preterm infant. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding The protein binding of lofexidine is determined to be moderate and it represents about 55% of the administered dose. |
| References |
[1]. The preclinical discovery of lofexidine for the treatment of opiate addiction. Expert Opin Drug Discov. 2014 Nov;9(11):1371-7. [2]. Lofexidine, an {alpha}2-receptor agonist for opioid detoxification. Ann Pharmacother. 2010 Feb;44(2):343-51. |
| Additional Infomation |
Lofexidine is a member of imidazoles, a dichlorobenzene, an aromatic ether and a carboxamidine. It has a role as an alpha-adrenergic agonist and an antihypertensive agent. Lofexidine is a non-opioid centrally acting alpha2-adrenergic receptor agonist that was first approved for the treatment of opioid withdrawal in the United Kingdom in 1992. It was first studied for use as an antihypertensive in 1980, but its researched was stopped as it was found less effective for the treatment of hypertension than clonidine. Lofexidine was then repurposed for the treatment of opioid withdrawal, as it was seen to be more economical and have fewer side effects than clonidine. Lofexidine was developed by US Woldmeds LLC and it was approved by the FDA on May 16, 2018. See also: Lofexidine Hydrochloride (has salt form). Drug Indication Lofexidine is indicated for mitigation of symptoms associated with acute withdrawal from opioids and for facilitation of the completion of opioid discontinuation treatment. It is the first non-opioid medication for the symptomatic management of opioid discontinuation. Opioid withdrawal syndrome is a debilitating manifestation of opioid dependence. This condition is extremely unpleasant lasting several days with some of the main features being abdominal pain, nausea, diarrhea, mydriasis, lacrimation, and piloerection. These symptoms are often observed after abrupt reductions in the opioid dose and can be resolved by re-administration of the opioid. FDA Label Mechanism of Action Lofexidine is a potent alpha2-adrenergic receptor agonist with some moderate agonistic affinity towards Alpha-1A adrenergic receptor and 5-HT1a, 5-HT7, 5HT2c and 5HT1d receptors. The alpha2-adrenergic receptor is normally targeted by norepinephrine and its activation inhibits the synthesis of cAMP which in turn leads to potassium efflux and suppression of neural firing and inhibition of norepinephrine release. All of this activity can reduce the heart rate, blood pressure, and attenuate sympathetic stress response. Opioids inhibit cAMP in the noradrenergic neurons and their discontinuation produces a rise in the level of cAMP. This will generate an increase in norepinephrine which is associated with the symptoms of withdrawal. The magnitude of the effect is augmented by chronic opioid use due to the compensatory mechanisms of continuous negative feedback. Therefore, chronic opioid use translates into an exacerbated production of cAMP and norepinephrine release. Lofexidine replaces the opioid-driven inhibition of cAMP production by activating the alpha2-adrenergic receptor and moderating the symptoms of opioid withdrawal. This effect is performed without interacting with opioid receptors which mediate other activities of opioid dependence or addiction. Pharmacodynamics In clinical trials, lofexidine presented more severe opioid withdrawal effects than observed with methadone. On the other hand, in clinical trials of methadone withdrawal, lofexidine effectively reduced withdrawal symptoms, especially hypotension. The clinical reports have also indicated that lofexidine presents a better outcome when used briefly. In phase 3 clinical trials, lofexidine was shown to generate a significantly higher completion rate of opioid discontinuation. Some pharmacological studies were performed and there were no off-target effects reported. |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (8.03 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (8.03 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (8.03 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.8591 mL | 19.2953 mL | 38.5907 mL | |
| 5 mM | 0.7718 mL | 3.8591 mL | 7.7181 mL | |
| 10 mM | 0.3859 mL | 1.9295 mL | 3.8591 mL |