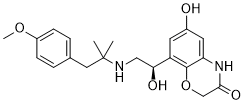
Olodaterol (formerly known as BI-1744; BI1744; STRIVERDI RESPIMAT) is a ultra-long acting beta-adrenoceptor agonist approved in 2014 for use as an inhalation for treating patients with chronic obstructive pulmonary disease (COPD) It was developed and manufactured by Boehringer-Ingelheim. For the treatment of chronic obstructive pulmonary disease, olastrol received FDA approval in 2014.
Physicochemical Properties
| Molecular Formula | C21H26N2O5 |
| Molecular Weight | 386.44 |
| Exact Mass | 386.184 |
| CAS # | 868049-49-4 |
| Related CAS # | Olodaterol hydrochloride; 869477-96-3 |
| PubChem CID | 11504295 |
| Appearance | Light yellow to khaki solid powder |
| Density | 1.3±0.1 g/cm3 |
| Boiling Point | 649.0±55.0 °C at 760 mmHg |
| Flash Point | 346.3±31.5 °C |
| Vapour Pressure | 0.0±2.0 mmHg at 25°C |
| Index of Refraction | 1.596 |
| LogP | 1.17 |
| Hydrogen Bond Donor Count | 4 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 28 |
| Complexity | 521 |
| Defined Atom Stereocenter Count | 1 |
| SMILES | [C@H](C1C=C(O)C=C2NC(COC=12)=O)(O)CNC(C)(C)CC1C=CC(OC)=CC=1 |
| InChi Key | COUYJEVMBVSIHV-SFHVURJKSA-N |
| InChi Code | InChI=1S/C21H26N2O5/c1-21(2,10-13-4-6-15(27-3)7-5-13)22-11-18(25)16-8-14(24)9-17-20(16)28-12-19(26)23-17/h4-9,18,22,24-25H,10-12H2,1-3H3,(H,23,26)/t18-/m0/s1 |
| Chemical Name | 6-hydroxy-8-[(1R)-1-hydroxy-2-[[1-(4-methoxyphenyl)-2-methylpropan-2-yl]amino]ethyl]-4H-1,4-benzoxazin-3-one |
| Synonyms | BI1744; BI-1744; BI 1744; Striverdi; Olodaterol |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | β2 adrenoceptor ( EC50 = 1 nM ) |
| ln Vitro | Olodaterol (0.001~10 nM; fibroblasts) inhibits the motility and proliferation induced by growth factors [2]. Fibroblasts treated with olotadrol (0.1–10 nM) prevent the signaling cascade from being phosphorylated when FGF is present [2]. Concentration-dependently, olapadaterol (0.001~1000 nM; 30 minutes; fibroblasts) raises intracellular cAMP. With a maximum efficacy of 70% at 10 nM, oledacaterol (0 to 10 nM; 30 min; fibroblasts) increased PICP in a concentration-dependent manner. Olodaterol is selective for the β2-AR receptor and has a subnanomolar affinity for it (pKi=9.14) when compared to the β1-AR and β3-AR subtypes [2]. |
| ln Vivo | Olodaterol (1 mg/kg; inhalation; day 21) attenuates TGF-β-induced pulmonary fibrosis and speeds up the weight return to control levels (day 21)[2]. After 0.5 hours, olodaterol (0.1 to 3 μg/kg; inhaled; 5h ) and olodaterol (0.3 and 0.6 μg/kg; inhaled; 24 hours)) induce about 60% anesthetic protection[3]. |
| Cell Assay |
Cell Line: Fibroblasts Concentration: 0.1~10 nM Result: Interfered with FGF-induced phosphorylation of signalling cascades. |
| Animal Protocol |
Lung fibrosis C57BL/6 mice 1 mg/mL Inhal.; 21 days |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Olodaterol reaches maximum plasma concentrations generally within 10 to 20 minutes following drug inhalation. In healthy volunteers, the absolute bioavailability of olodaterol following inhalation was estimated to be approximately 30%, whereas the absolute bioavailability was below 1% when given as an oral solution. Thus, the systemic availability of olodaterol after inhalation is mainly determined by lung absorption, while any swallowed portion of the dose only negligibly contributes to systemic exposure. Following intravenous administration of [14C]-labeled olodaterol, 38% of the radioactive dose was recovered in the urine and 53% was recovered in feces. The amount of unchanged olodaterol recovered in the urine after intravenous administration was 19%. Following oral administration, only 9% of olodaterol and/or its metabolites was recovered in urine, while the major portion was recovered in feces (84%). The volume of distribution is high (1110 L), suggesting extensive distribution into tissue. Total clearance of olodaterol in healthy volunteers is 872 mL/min, and renal clearance is 173 mL/min. Metabolism / Metabolites Olodaterol is substantially metabolized by direct glucuronidation and by O-demethylation at the methoxy moiety followed by conjugation. Of the six metabolites identified, only the unconjugated demethylation product binds to beta2-receptors. This metabolite, however, is not detectable in plasma after chronic inhalation of the recommended therapeutic dose. Cytochrome P450 isozymes CYP2C9 and CYP2C8, with negligible contribution of CYP3A4, are involved in the O-demethylation of olodaterol, while uridine diphosphate glycosyl transferase isoforms UGT2B7, UGT1A1, 1A7, and 1A9 were shown to be involved in the formation of olodaterol glucuronides. Biological Half-Life The terminal half-life following intravenous administration is 22 hours. The terminal half-life following inhalation in contrast is about 45 hours, indicating that the latter is determined by absorption rather than by elimination processes. |
| Toxicity/Toxicokinetics |
Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Although no published data exist on the use of olodaterol during lactation, data from the related drug, terbutaline, indicate that very little is expected to be excreted into breastmilk. The authors of several reviews agree that use of inhaled bronchodilators is acceptable during breastfeeding because of the low bioavailability and maternal serum levels after use. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding In vitro binding of olodaterol to human plasma proteins is independent of concentration and is approximately 60%. |
| References |
[1]. Design, synthesis and biological evaluation of 8-(2-amino-1-hydroxyethyl)-6-hydroxy-1,4-benzoxazine-3(4H)-one derivatives as potent β2-adrenoceptor agonists. Bioorg Med Chem. 2020;28(1):115178. [2]. Olodaterol shows anti-fibrotic efficacy in in vitro and in vivo models of pulmonary fibrosis. Br J Pharmacol. 2017;174(21):3848-3864. [3]. Pharmacological characterization of olodaterol, a novel inhaled beta2-adrenoceptor agonist exerting a 24-hour-long duration of action in preclinical models [published correction appears in J Pharmacol Exp Ther. 2013 Jul;346(1):161]. J Pharmacol Exp Ther. 2010;334(1):53-62. |
| Additional Infomation |
Olodaterol is a member of the class of benzoxazine that is 6-hydroxy-1,4-benzoxazin-3-one in which the hydrogen at position 4 is replaced by a (1R)-1-hydroxy-2-{[1-(4-methoxyphenyl)-2-methylpropan-2-yl]amino}ethyl group. Used (as its hydrochloride salt) for long-term treatment of airflow obstruction in patients with chronic obstructive pulmonary disease including chronic bronchitis and/or emphysema. It has a role as a beta-adrenergic agonist and a bronchodilator agent. It is a benzoxazine, a member of phenols, an aromatic ether, a secondary alcohol and a secondary amino compound. It is a conjugate base of an olodaterol(1+). Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma. Olodaterol is a beta2-Adrenergic Agonist. The mechanism of action of olodaterol is as an Adrenergic beta2-Agonist. See also: Olodaterol Hydrochloride (active moiety of). Drug Indication Olodaterol is indicated for use in chronic obstructive pulmonary disease (COPD), including chronic bronchitis and/or emphysema. It is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma. FDA Label Mechanism of Action Olodaterol is a long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~77 mg/mL (~199.3 mM) Ethanol: ~40 mg/mL |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5877 mL | 12.9386 mL | 25.8772 mL | |
| 5 mM | 0.5175 mL | 2.5877 mL | 5.1754 mL | |
| 10 mM | 0.2588 mL | 1.2939 mL | 2.5877 mL |