Metoprolol Tartrate (CGP2175E; CGP-2175E; Lanoc; Lopressor; Metomerck; Metop; Selopral; Ritmolol), the tartrate salt of Metoprolol, is a potent and cardioselective β1 receptor blocker with antihypertensive effects. It is a medication for high blood pressure and heart failure.
Physicochemical Properties
| Molecular Formula | C34H56N2O12 | |
| Molecular Weight | 684.81 | |
| Exact Mass | 684.38 | |
| Elemental Analysis | C, 59.63; H, 8.24; N, 4.09; O, 28.03 | |
| CAS # | 56392-17-7 | |
| Related CAS # | Metoprolol; 51384-51-1; Metoprolol succinate; 98418-47-4; Metoprolol-d7 hydrochloride; 1219798-61-4; Metoprolol-d6 tartrate; Metoprolol succinate;98418-47-4;Metoprolol-d7 hydrochloride;1219798-61-4;Metoprolol tartrate;56392-17-7;Metoprolol-d7;959787-96-3;(R)-Metoprolol-d7;1292907-84-6;(S)-Metoprolol-d7;1292906-91-2;Metoprolol-d5;959786-79-9; 56392-18-8 (HCl); 80274-67-5 (fumarate) | |
| PubChem CID | 441308 | |
| Appearance | White to off-white crystalline powder | |
| Boiling Point | 398.6ºC at 760 mmHg | |
| Melting Point | 120ºC | |
| Flash Point | 194.9ºC | |
| LogP | 1.885 | |
| Hydrogen Bond Donor Count | 8 | |
| Hydrogen Bond Acceptor Count | 14 | |
| Rotatable Bond Count | 21 | |
| Heavy Atom Count | 48 | |
| Complexity | 349 | |
| Defined Atom Stereocenter Count | 2 | |
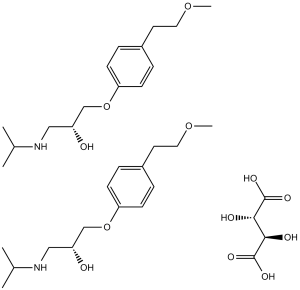
| SMILES | O(C1C([H])=C([H])C(=C([H])C=1[H])C([H])([H])C([H])([H])OC([H])([H])[H])C([H])([H])C([H])(C([H])([H])N([H])C([H])(C([H])([H])[H])C([H])([H])[H])O[H].O(C1C([H])=C([H])C(=C([H])C=1[H])C([H])([H])C([H])([H])OC([H])([H])[H])C([H])([H])C([H])(C([H])([H])N([H])C([H])(C([H])([H])[H])C([H])([H])[H])O[H].O([H])[C@@]([H])(C(=O)O[H])[C@]([H])(C(=O)O[H])O[H] |
|
| InChi Key | YGULWPYYGQCFMP-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/2C15H25NO3.C4H6O6/c2*1-12(2)16-10-14(17)11-19-15-6-4-13(5-7-15)8-9-18-3;5-1(3(7)8)2(6)4(9)10/h2*4-7,12,14,16-17H,8-11H2,1-3H3;1-2,5-6H,(H,7,8)(H,9,10) | |
| Chemical Name | 2,3-dihydroxybutanedioic acid;1-[4-(2-methoxyethyl)phenoxy]-3-(propan-2-ylamino)propan-2-ol | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | β-adrenergic receptor |
| ln Vitro | Metoprolol (0-1000 μg/mL; 24-72 h) exhibits dose- and time-dependent cytotoxicity on MOLT-4 and U937 cells[3]. |
| ln Vivo |
Metoprolol (2.5 mg/kg/h; infusion; 11 weeks) decreases atherosclerosis and proinflammatory cytokines in ApoE-/- mice[1]. Metoprolol (15 mg/kg/q12h; i.e., 5 days) exhibits anti-viral and anti-inflammatory properties in a murine model of viral myocarditis caused by the coxsackievirus B3[2]. Metoprolol (2.5 mg/kg; intravenously; three bolus injections) inhibits myocardial apoptosis and significantly reduces the expression of activated caspase-9 protein in coronary microembolization (CME) rats[4]. Carvedilol had a stronger effect than metoprolol in reducing the pathological scores of VMC induced by CVB3. Both carvedilol and metoprolol reduced the levels of cTn-I, but the effect of carvedilol was stronger. Carvedilol and metoprolol decreased the levels of myocardial pro-inflammatory cytokines and increased the expression of anti-inflammatory cytokine, with the effects of carvedilol being stronger than those of metoprolol. Carvedilol had a stronger effect in reducing myocardial virus concentration compared with metoprolol. Carvedilol was stronger than metoprolol in decreasing the levels of myocardial phosphorylated p38MAPK.Conclusions: In conclusion, carvedilol was more potent than metoprolol in ameliorating myocardial lesions in VMC, probably due to its stronger modulation of the balance between pro- and anti-inflammatory cytokines by inhibiting the activation of p38MAPK pathway through β1- and β2-adrenoreceptors.[2] The echocardiographic parameters of left ventricular function were significantly decreased in the CME group compared with the control group (P<0.05); however, the metoprolol group and ZLF group showed significantly improved cardiac function compared with CME alone (P<0.05). Compared with the control group, the myocardial apoptosis rate and the levels of activated caspase-9 and -3 increased significantly in the CME group (P<0.05). Again, these effects were ameliorated by metoprolol and ZLF (P<0.05).Conclusions: The present study demonstrates that metoprolol and ZLF can protect the rat myocardium during CME by inhibiting apoptosis and improving cardiac function, likely by inhibiting apoptosis/ mitochondrial apoptotic pathway. These results suggest that antiapoptotic therapies may be useful in treating CME.[3] |
| Cell Assay |
Cell Line: U937 and MOLT-4 cells Concentration: 1, 10, 50, 100, 500 and 1000 μg/mL Incubation Time: 24, 48 and 72 h Result: Significantly reduced the viability of MOLT-4 and U937 cells at 1000 μg/mL (3740.14µM) concentration after 48 hours of incubation; similarly, after 72 hours, the viability of MOLT4 cells at ≥100 μg/ml (≥374.01µM) concentrations and U937 cells at ≥500 μg/ml (≥1870.07µM) concentrations was observed. |
| Animal Protocol |
Male ApoE-/- mice 2.5 mg/kg/h Via osmotic minipumps, 11 weeks A total of 116 Balb/c mice were included in this study. Ninety-six mice were inoculated intraperitoneally with CVB3 to induce VMC. The CVB3 inoculated mice were evenly divided into myocarditis group (n=32), carvedilol group (n=32) and metoprolol group (n=32). Twenty mice (control group) were inoculated intraperitoneally with normal saline. Hematoxylin and eosin staining and histopathologic scoring were used to investigate the effects of carvedilol and metoprolol on myocardial histopathologic changes on days 3 and 5. In addition, serum cTn-I levels, cytokine levels and virus titers were determined using chemiluminescence immunoassay, enzyme-linked immunosorbent assay and plaque assay, respectively, on days 3 and 5. Finally, the levels of phosphorylated p38MAPK were studied using immunohistochemical staining and Western blotting on day 5.[2] Forty rats were randomly divided into four groups (n=10 each): a sham operation (control) group, CME plus saline (CME) group, CME plus metoprolol (metoprolol) group and caspase-9 inhibitor Z-LEHD-FMK (ZLF) group. CME was induced by injecting 3000 polyethylene microspheres (42 μm diameter) into the left ventricle during a 10 s occlusion of the ascending aorta. Echocardiography, terminal deoxynucleotidyl transferase dUTP nick end labelling and Western blotting were used to evaluate cardiac function, apoptosis and activation of caspase-9/caspase-3, respectively, 6 h after CME.[3] A few studies in animals and humans suggest that metoprolol (β1-selective adrenoceptor antagonist) may have a direct antiatherosclerotic effect. However, the mechanism behind this protective effect has not been established. The aim of the present study was to evaluate the effect of metoprolol on development of atherosclerosis in ApoE(-/-) mice and investigate its effect on the release of proinflammatory cytokines. Male ApoE(-/-) mice were treated with metoprolol (2.5 mg/kg/h) or saline for 11 weeks via osmotic minipumps. Atherosclerosis was assessed in thoracic aorta and aortic root. Total cholesterol levels and Th1/Th2 cytokines were analyzed in serum and macrophage content in lesions by immunohistochemistry. Metoprolol significantly reduced atherosclerotic plaque area in thoracic aorta (P < 0.05 versus Control). Further, metoprolol reduced serum TNFα and the chemokine CXCL1 (P < 0.01 versus Control for both) as well as decreasing the macrophage content in the plaques (P < 0.01 versus Control). Total cholesterol levels were not affected. In this study we found that a moderate dose of metoprolol significantly reduced atherosclerotic plaque area in thoracic aorta of ApoE(-/-) mice. Metoprolol also decreased serum levels of proinflammatory cytokines TNFα and CXCL1 and macrophage content in the plaques, showing that metoprolol has an anti-inflammatory effect.[1] |
| ADME/Pharmacokinetics |
Absorption When metoprolol is administered orally, it is almost completely absorbed in the gastrointestinal tract. The maximum serum concentration is achieved 20 min after intravenous administration and 1-2 hours after oral administration. The bioavailability of metoprolol is of 100% when administered intravenously and when administered orally it presents about 50% for the tartrate derivative and 40% for the succinate derivative. The absorption of metoprolol in the form of the tartrate derivative is increased by the concomitant administration of food. Route of Elimination Metoprolol is mainly excreted via the kidneys. From the eliminated dose, less than 5% is recovered unchanged. Volume of Distribution The reported volume of distribution of metoprolol is 4.2 L/kg. Due to the characteristics of metoprolol, this molecule is able to cross the blood-brain barrier and even 78% of the administered drug can be found in cerebrospinal fluid. Clearance The reported clearance rate on patients with normal kidney function is 0.8 L/min. In cirrhotic patients, the clearance rate changes to 0.61 L/min. Plasma levels following oral administration of conventional metoprolol tablets, however, approximate 50% of levels following intravenous adminsitration, indicating about 50% first-pass metabolism... Elimination is mainly by biotransformation in the liver. View More
Metoprolol tartrate is rapidly and almost completely absorbed from the GI tract; absorption of a single oral dose of 20-100 mg is complete in 2.5-3 hours. After an oral dose, about 50% of the drug administered as conventional tablets appears to undergo first-pass metabolism in the liver. Bioavailability of orally administered metoprolol tartrate increases with increased doses, indicating a possible saturable disposition process of low capacity such as tissue binding in the liver. Steady-state oral bioavailability of extended-release tablets of metoprolol succinate given once daily at dosages equivalent to 50-400 mg of metoprolol tartrate is about 77% of that of conventional tablets at corresponding dosages given once daily or in divided doses. Food does not appear to affect bioavailability of metoprolol succinate extended-release tablets. Following a single oral dose as conventional tablets, metoprolol appears in the plasma within 10 minutes and peak plasma concentrations are reached in about 90 minutes. When metoprolol tartrate conventional tablets are administered with food rather than on an empty stomach, peak plasma concentrations are higher and the extent of absorption of the drug is increased. Following oral administration of metoprolol succinate as extended-release tablets, peak plasma metoprolol concentrations are aobut 25-50% of those attained after administration of metoprolol tartrate conventional tablets given once daily or in divided doses. Time to peak concentration is longer with extended-release tablets, with peak plasma coentrations being reached in about 7 hours following administration of such tablets. Plasma concentrations attained 1 hour after an oral dose are linearly related to metoprolol tartrate doses ranging from 50-400 mg as conventional tablets.
Metabolism / Metabolites Metoprolol goes through significant first-pass hepatic metabolism which covers around 50% of the administered dose. The metabolism of metoprolol is mainly driven by the activity of CYP2D6 and to a lesser extent due to the activity of CYP3A4. The metabolism of metoprolol is mainly represented by reactions of hydroxylation and O-demethylation. Metoprolol does not inhibit or enhance its own metabolism. Three main metabolites of the drug are formed by oxidative deamination, O-dealkylation with subsequent oxidation, and aliphatic hydroxylation; these metabolites account for 85% of the total urinary excretion of metabolites. The metabolites apparently do not have appreciable pharmacologic activity. The rate of hydroxylation, resulting in alpha-hydroxymetoprolol, is genetically determined and is subject to considerable interindividual variation. Poor hydroxylators of metoprolol have increased areas under the plasma concentration-time curves, prolonged elimination half-lives (about 7.6 hours), higher urinary concentrations of unchanged drug, and negligible urinary concentrations of alpha-hydroxymetoprolol compared with extensive hydroxylators. Beta-adrenergic blockade of exercise-induced tachycardia persists for at least 24 hours after administration of a single 200-mg oral dose of metoprolol tartrate in poor hydroxylators. Controlled studies have shown that debrisoquine oxidation phenotype is a major determinant of the metabolism, pharmacokinetics and some of the pharmacological actions of metoprolol. The poor metabolizer phenotype is associated with increased plasma drug concentrations, a prolongation of elimination half-life and more intense and sustained beta blockade. Phenotypic differences have also been observed in the pharmacokinetics of the enantiomers of metoprolol. In vivo and in vitro studies have identified some of the metabolic pathways which are subject to the defect, that is alpha-hydroxylation and O-demethylation. PMID:2868819 Metropolol is a racemic mixture of R-and S-enantiomers, and is primarily metabolized by CYP2D6. Biological Half-Life The immediate release formulations of metoprolol present a half-life of about 3-7 hours. The plasma half-life ranges from approximately 3 to 7 hours. |
| Toxicity/Toxicokinetics |
Hepatotoxicity
Metoprolol therapy has been associated with a low rate of mild-to-moderate elevations of serum aminotransferase levels which are usually asymptomatic and transient and resolve even with continuation of therapy. A few instances of clinically apparent, acute liver injury attributable to metoprolol have been reported. In view of its wide scale use, metoprolol induced liver injury is exceedingly rare. The typical liver injury associated with beta-blockers has a latency to onset of 2 to 12 weeks and a hepatocellular pattern of liver enzyme. Symptoms of hypersensitivity (rash, fever, eosinophilia) and autoantibody formation have not been reported. Reported cases due to metoprolol have included cases of acute liver failure, but ultimately all were self-limiting and resolved fairly rapidly once once drug was stopped. Likelihood score: D (possible rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Because of the low levels of metoprolol in breastmilk, amounts ingested by the infant are small and would not be expected to cause any adverse effects in breastfed infants. Studies on the use of metoprolol during breastfeeding have found no adverse reactions in breastfed infants. Monitor breastfed infants for symptoms of beta blockade such as bradycardia and listlessness due to hypoglycemia. ◉ Effects in Breastfed Infants A study of mothers taking beta-blockers during nursing found a numerically, but not statistically significant increased number of adverse reactions in those taking any beta-blocker. Although the ages of infants were matched to control infants, the ages of the affected infants were not stated. Of 6 mothers taking metoprolol, none reported adverse effects in her breastfed infant. ◉ Effects on Lactation and Breastmilk Relevant published information on the effects of beta-blockade or metoprolol during normal lactation was not found as of the revision date. A study in 6 patients with hyperprolactinemia and galactorrhea found no changes in serum prolactin levels following beta-adrenergic blockade with propranolol. View More
◈ What is metoprolol?
Interactions The effect of verapamil coadministration on the hepatic first pass clearance of metoprolol was investigated in dogs. Plasma concentration-time course of metoprolol enantiomers and urinary recovery of oxidative metabolites were determined after a single iv (0.51 mg/kg) and an oral (1.37 mg/kg) dose of deuterium labeled pseudoracemic metoprolol, with or without concomitant administration of racemic verapamil (3 mg/kg). Verapamil inhibited both the systemic and oral clearance of metoprolol by about 50-70%. The first pass effect of metoprolol was completely abolished after coadministration of verapamil, reflecting a marked alteration in the degree of hepatic extraction of metoprolol from intermediate to low. The hepatic clearance of metoprolol was slightly (S)-enantioselective (R/S ratio = 0.89 + or - 0.04) in control dogs. Inhibition of hepatic clearance of metoprolol by verapamil was selective towards (S)-metoprolol, such that the enantioselectivity in hepatic clearance toward (S)-metoprolol disappeared following verapamil coadministration (R/S ratio = 1.01 + or - 0.05). Urinary metabolite profiles indicated that O-demethylation and N-dealkylation were the major pathways of oxidative metabolism in the dog. alpha-Hydroxymetoprolol was a minor metabolite in urine. N-Dealkylation showed a strong preference for (S)-metoprolol, whereas O-demethylation and alpha-hydroxylation exhibited a modest selectivity toward (R)-metoprolol; hence, the slight (S)-enantioselectivity in the overall hepatic clearance. Comparison of metoprolol metabolite formation clearances in the absence or presence of verapamil coadministration showed that all three oxidative pathways were inhibited by 60-80%. The greater inhibition of hepatic clearance observed with (S)-metoprolol as compared to (R)-metoprolol was attributed to a significant (S)-enantioselective inhibition in the O-demethylation of metoprolol by verapamil. PMID:1687016 The interaction between metoprolol and bromazepam and lorazepam was studied in 12 healthy male volunteers aged 21-37 years. Metoprolol had no significant effect on the pharmacokinetics of bromazepam or lorazepam. However, bromazepam area under the curve was 35% higher in the presence of metoprolol. Bromazepam enhanced the effect of metoprolol on systolic blood pressure but not on diastolic blood pressure or pulse rate. Lorazepam had no effect on either blood pressure or pulse. Metoprolol did not enhance the effect of bromazepam on the psychomotor tests used in this study. Metoprolol caused a small increase in critical flicker fusion threshold with lorazepam but had no effect on the other tests. Lorazepam (2 mg) was more potent than bromazepam (6 mg) in the doses used in this study. The interaction of metoprolol with bromazepam and lorazepam is unlikely to be of clinical significance. No change in dose is necessary when using these drugs together. Protein Binding Metoprolol is not highly bound to plasma proteins and only about 11% of the administered dose is found bound. It is mainly bound to serum albumin. |
| References |
[1]. Metoprolol reduces proinflammatory cytokines and atherosclerosis in ApoE-/- mice. Biomed Res Int. 2014;2014:548783. [2]. Carvedilol has stronger anti-inflammation and anti-virus effects than metoprolol in murine model with coxsackievirus B3-induced viral myocarditis. Gene. 2014 Sep 1;547(2):195-201. [3]. Cytotoxicity of Metoprolol on Leukemic Cells in Vitro. IJBC 2018; 10(4): 124-129. [4]. Effect of metoprolol on myocardial apoptosis and caspase-9 activation after coronary microembolization in rats. Exp Clin Cardiol. 2013 Spring;18(2):161-5. |
| Additional Infomation |
Metoprolol tartrate is a member of phenols and an alcohol. Metoprolol Tartrate is the tartrate salt form of metoprolol, a cardioselective competitive beta-1 adrenergic receptor antagonist with antihypertensive properties and devoid of intrinsic sympathomimetic activity. Metoprolol tartrate antagonizes beta 1-adrenergic receptors in the myocardium, thereby reducing the rate and force of myocardial contraction, and consequently a diminished cardiac output. This agent may also reduce the secretion of renin with subsequent reduction in levels of angiotensin II thus decreasing sympathetic activation, including vasoconstriction, aldosterone secretion. A selective adrenergic beta-1 blocking agent that is commonly used to treat ANGINA PECTORIS; HYPERTENSION; and CARDIAC ARRHYTHMIAS. See also: Metoprolol (has active moiety); Hydrochlorothiazide; metoprolol tartrate (component of); Chlorthalidone; metoprolol tartrate (component of). |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.4603 mL | 7.3013 mL | 14.6026 mL | |
| 5 mM | 0.2921 mL | 1.4603 mL | 2.9205 mL | |
| 10 mM | 0.1460 mL | 0.7301 mL | 1.4603 mL |