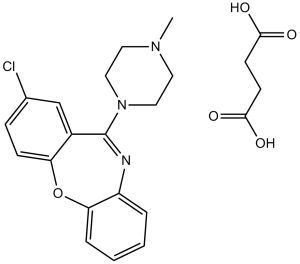
Loxapine Succinate (Loxapac; Adasuve; Cloxazepin; Daxolin; Oxilapine), the succinate salt of Loxapine which is a dibenzoxazepine anti-psychotic drug, is a potent D2DR/D4DR inhibitor as well as serotonergic receptor antagonist. It is a common antipsychotic medication that is mostly used to treat schizophrenia.
Physicochemical Properties
| Molecular Formula | C22H24CLN3O5 | |
| Molecular Weight | 445.9 | |
| Exact Mass | 445.14 | |
| Elemental Analysis | C, 59.26; H, 5.43; Cl, 7.95; N, 9.42; O, 17.94 | |
| CAS # | 27833-64-3 | |
| Related CAS # | Loxapine; 1977-10-2; Loxapine hydrochloride; 54810-23-0; Loxapine-d8; 1189455-63-7 | |
| PubChem CID | 71399 | |
| Appearance | White to off-white solid powder | |
| Boiling Point | 458.6ºC at 760mmHg | |
| Melting Point | 150-152°C | |
| Flash Point | 231.1ºC | |
| LogP | 3.018 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 7 | |
| Rotatable Bond Count | 4 | |
| Heavy Atom Count | 31 | |
| Complexity | 542 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | ClC1C([H])=C([H])C2=C(C=1[H])C(=NC1=C([H])C([H])=C([H])C([H])=C1O2)N1C([H])([H])C([H])([H])N(C([H])([H])[H])C([H])([H])C1([H])[H].O([H])C(C([H])([H])C([H])([H])C(=O)O[H])=O |
|
| InChi Key | YQZBAXDVDZTKEQ-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C18H18ClN3O.C4H6O4/c1-21-8-10-22(11-9-21)18-14-12-13(19)6-7-16(14)23-17-5-3-2-4-15(17)20-18;5-3(6)1-2-4(7)8/h2-7,12H,8-11H2,1H3;1-2H2,(H,5,6)(H,7,8) | |
| Chemical Name | butanedioic acid;8-chloro-6-(4-methylpiperazin-1-yl)benzo[b][1,4]benzoxazepine | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | human 5-HT2; Human D4 Receptor; Human D1 Receptor; Human D2Receptor | |
| ln Vitro |
|
|
| ln Vivo |
|
|
| Enzyme Assay |
Receptor binding assays - dopamine, 5-HT2, NMDA receptors [1] To perform the receptor binding assays, 0.8 nM of [3H] SCH23390 (Di receptor antagonist), 0.5 nM [3H] spiroperidol (D2 and D4 receptor antagonist), 0.5 nM of [3H] ketanserin (5-HT2 receptor antagonist), and 2.0 nM [3H] MK801 (NMDA receptor antagonist) were incubated with 150 )ig of membrane proteins in a final volume of 1 ml. Nonspecific binding was determined in parallel assays in the presence of 1 jM (+) butaclamol (D2 and D4 assays), 10 jM cis-flupenthixol (Di assays), 2 ,uM methysergide (5-HT2 assays) and 50 jM MK801 (NMDA assays). Assays using [3H] spiroperidol also included 50 nM ketanserin to occlude the presence ofserotonergic sites. For the competition experiments, varying concentrations of loxapine were included in the assay tubes. Incubations forthe Di, D2, 5-HT2 andNMDA receptors were performed at 25°C for 90 min, 25°C for 60 min, 37°C for 15 min and 25°C for 120 min, respectively. D4 receptor binding assays with COS cells were incubated at 22°C for 120 min using the cell binding buffer described in the membrane preparation section. At the end ofthe incubation, the bound and free ligands were separated by rapid filtration on Whatman GF/B filters, which were washed 3 times with 5 ml ofcold filtration buffer: (50 mM Tris-HCL, 1.0 mM EDTA, pH 7.4) for the [3H] spiroperidol and [3H] SCH23390 assays, (50 mM Tris-HCL, pH 7.4) for [3H] ketanserin assays, and (10 mM HEPES, 1 mM EDTA, pH 7.4) for [3H] MK80 1 assays. Bound radioactivity was measured using a Beckman Scintillation Counter (model LS 5000TA).[1] |
|
| Cell Assay | The cytokines IL-1beta and IL-2 are released from activated glial cells in the central nervous system and they are able to enhance catecholaminergic neurotransmission. There is no data concerning influence of antipsychotics on glial cell activity. Antipsychotics reaching the brain act not only on neurons but probably also on glial cells. The aim of this study was to evaluate the effect of chlorpromazine and loxapine on release of IL-1beta and IL-2 by mixed glial and microglial cell cultures. Chlorpromazine in concentrations 2 and 20 muM, and loxapine 0.2, 2 and 20 microM reduced IL-1beta secretion by LPS-activated mixed glia cultures after 1 and 3 days of exposure. Chlorpromazine in concentrations of 0.2, 2 and 20 microM reduced the IL-2 secretion in mixed glial cultures after 3 days of exposure. Loxapine in concentrations of 0.2, 2 and 20 microM reduced IL-2 secretion in mixed glia cultures after 1 and 3 days of exposure, and additionally loxapine decreased IL-1beta and IL-2 secretion in LPS-induced microglia cultures in concentrations of 2, 10 and 20 muM. Quinpirole-a D2 dopaminergic agonist increased LPS-induced IL-1beta and IL-2 secretion in mixed glia cultures only in the highest dose of 20 microM. These findings suggest the absence of functional dopamine receptors on cortical microglial cells. Mixed glia cultures deprived of microglia (by shaking and incubating with L-leucine methyl ester) did not release IL-1beta and IL-2. This observation suggests that microglia can be a source of assessed cytokines. Results of the present study support the view that antipsychotics act not only on neurons but also on glial cells. However, the clinical significance of these observations still remains unclear[2]. | |
| Animal Protocol |
Adult male Wistar rats (150-175 g) 5 mg/kg Intraperitoneal injection, daily for 4 or 10 weeks Loxapine (0.3 mg/kg s.c.), olanzapine (10 mg/kg s.c.) and SCH 23390 (R-(+)-chloro-2, 3, 4, 5-tetrahydro-3-methyl-5-phenyl-1-H-3-benzazepine; 1 mg/kg, s.c.), but not clozapine (10 mg/kg, s.c.), induced catalepsy in rats. Co-administration of clozapine (1, 3 and 10 mg/kg s.c.) dose-dependently inhibited loxapine-induced catalepsy. Clozapine (10 mg/kg s.c.) also prevented the induction of catalepsy by olanzapine. In addition, clozapine abolished the catalepsy induced by loxapine when it was administered after the response had fully developed. In contrast, the duration of SCH 23390-induced catalepsy was prolonged by clozapine, indicating that its anti-catalepsy effects against olanzapine and loxapine are unlikely to be caused by muscle relaxation, sedation or stimulation. Since SCH 23390-induced catalepsy is reported to be blocked by scopolamine, dizocilpine (MK-801) or 8-hydroxy-dipropylamino-tetralin, it is unlikely that muscarinic blockade, NMDA ion channel blockade and 5-HT1A receptor agonism, respectively, are involved in clozapine's action, but the mechanism by which clozapine exerts this anti-cataleptic effect remains unknown.[4] |
|
| ADME/Pharmacokinetics |
Absorption Systemic bioavailability of the parent drug was only about one third that after an equivalent intramuscular dose (25 mg base) in male volunteers Route of Elimination Metabolites are excreted in the urine in the form of conjugates and in the feces unconjugated. Animal studies with radioactive drug indicate that loxapine and/or its metabolites are widely distributed in body tissues with highest concentrations in brain, lungs, heart, liver, and pancreas. The drug appears in the CSF. Loxapine is rapidly and almost completely absorbed from the GI tract. The drug is also almost completely absorbed following IM administration. RAPIDLY & ALMOST COMPLETELY ABSORBED FROM GI TRACT. PEAK LOXAPINE SERUM LEVELS /WITHIN 2 HR, RANGE FROM 0.006 TO 0.013 MCG/ML AFTER/ 25 MG ORAL DOSE...MAJOR /ACTIVE/ METABOLITE IN SERUM IS 8-HYDROXYLOXAPINE /MAX CONCN 0.012-0.038 MCG/ML WITHIN 2-4 HR AFTER ORAL LOXAPINE. HUMAN/ LOXAPINE AND/OR METABOLITES...WIDELY DISTRIBUTED IN BODY TISSUES...HIGHEST CONCN IN BRAIN, LUNGS, HEART, LIVER, & PANCREAS...APPEARS IN CSF...CROSSES PLACENTA...IN MILK OF NURSING MOTHERS /ANIMALS, RADIOACTIVE DRUG/ METABOLITES /7- & 8-HYDROXY-, 7- & 8-HYDROXYDESMETHYLLOXAPINE; N-OXIDES OF LOXAPINE, 7- & 8-HYDROXYLOXAPINE/ EXCRETED IN URINE & FECES. LITTLE OR NO UNMETABOLIZED DRUG...FOUND...METABOLITES /PRIMARILY GLUCURONIDE OR SULFATE CONJUGATES IN URINE, PRIMARILY UNCONJUGATED IN FECES. HUMAN, ORAL/ View More
Metabolism / Metabolites
Biological Half-Life Oral-4 hours SERUM LEVELS OF LOXAPINE & METABOLITES DECLINE IN BIPHASIC MANNER. HALF-LIFE DURING 1ST PHASE...5 HR...DURING 2ND PHASE...19 HR. /AFTER SINGLE 25 MG ORAL DOSE, SEDATIVE EFFECT BEGINS IN 20-30 MIN; PEAK EFFECT WITHIN 1.5-3 HR; DURATION APPROX 12 HR. HUMAN/ |
|
| References |
[1]. J Psychiatry Neurosci . 1996 Jan;21(1):29-35. [2]. Eur Neuropsychopharmacol . 2005 Jan;15(1):23-30. [3]. Psychiatry Res . 1984 Aug;12(4):277-85. [4]. Naunyn Schmiedebergs Arch Pharmacol . 1997 Mar;355(3):361-4. |
|
| Additional Infomation |
Loxapine Succinate is the succinate salt form of loxapine, a tricyclic dibenzoxazepine antipsychotic agent with antiemetic, sedative, anticholinergic, and antiadrenergic actions. Loxapine succinate exerts its actions by blocking the dopamine receptors at postsynaptic receptor sites in the limbic system, cortical system and basal ganglia, thereby reducing the hallucinations and delusions that are associated with schizophrenia. This agent also exerts extrapyramidal side effects. An antipsychotic agent used in schizophrenia. See also: Loxapine (has active moiety). |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.61 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.61 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.61 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2427 mL | 11.2133 mL | 22.4266 mL | |
| 5 mM | 0.4485 mL | 2.2427 mL | 4.4853 mL | |
| 10 mM | 0.2243 mL | 1.1213 mL | 2.2427 mL |