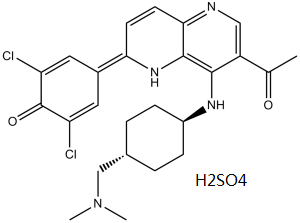
OTSSP167 sulfate (OTS167 HCl), the sulfate salt of OTSSP-167 (OTS-167), is an orally bioavailable inhibitor of MELK (maternal embryonic leucine zipper kinase) with IC50 of 0.41 nM. MELK may be a promising target for GC treatment.
Physicochemical Properties
| Related CAS # | 1431697-89-0; 1431698-10-0 |
| Appearance | Typically exists as solid at room temperature |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | MELK (IC50 = 0.41 nM) |
| ln Vitro | OTSSP167 inhibits the growth of A549 (lung), T47D (breast), DU4475 (breast), 22Rv1 (prostate) and HT1197 (bladder) cancer cells with IC50 values of 6.7, 4.3, 2.3, 6.0 and 97 nM, respectively[1]. OTSSP167 can abrogate the mitotic checkpoint, disrupt MCC and MCC-APC/C interaction in MCF7 cells. OTSSP167 causes GFP-MELK localization to cell cortex in prometaphase cells[2]. OTSSP167 is a MELK selective inhibitor, exhibits a strong in vitro activity, conferring an IC50 of 0.41 nM[3]. |
| ln Vivo | Growth suppressive effect of OTSSP167 in xenograft mouse model [1] We subsequently investigated in vivo anti-tumor effect of OTSSP167 by a xenograft model using MDA-MB-231 cells (MELK-positive, triple-negative breast cancer cells). The compound was administered to mice bearing xenografts for 14 days after the tumor size reached about 100 mm3. The tumor size was measured as a surrogate marker of drug response (tumor growth inhibition (TGI)). Intravenous administration of OTSSP167 at 20 mg/kg once every two days resulted in TGI of 73% (Fig 3A). Since the bioavailability of this compound was expected to be very high (data not shown), we attempted oral administration of this compound. The oral administration at 10 mg/kg once a day revealed TGI of 72% (Fig 3B). Due to the strong growth-suppressive effect on various cancer cell lines, we further investigated in vivo growth-suppressive effect using cancer cell lines of other types and found significant tumor growth suppression by OTSSP167 for multiple cancer types in dose-dependent manners with no or a little body-weight loss (Fig 3 and Supplementary Fig. S1). For example, mice carrying A549 (lung cancer) xenografts that were treated with 1, 5, and 10 mg/kg once a day of OTSSP167 by intravenous administration revealed TGI of 51, 91, and 108%, respectively (Fig 3C) and those by oral administration of 5 and 10 mg/kg once a day revealed TGI of 95 and 124%, respectively (Fig 3D). In addition, we examined DU145 (prostate cancer) and MIAPaCa-2 (pancreatic cancer) xenograft models by oral administration of 10 mg/kg once a day, and observed TGI of 106 and 87%, respectively (Fig 3E and F). To further validate the MELK-specific in vivo tumor suppressive effect, we examined PC-14 lung cancer cells in which MELK expression was hardly detectable (Fig 3G). Oral administration of 10 mg/kg OTSSP167 once a day for 14 days showed no tumor growth suppressive effect on PC-14 xenografts (Fig 3H), further supporting the MELK-dependent antitumor activity of OTSSP167. |
| References |
[1]. Development of an orally-administrative MELK-targeting inhibitor that suppresses the growth of various types of human cancer. Oncotarget. 2012 Dec;3(12):1629-40. [2]. Maternal embryonic leucine zipper kinase serves as a poor prognosis marker and therapeutic target in gastric cancer. Oncotarget. 2016 Feb 2;7(5):6266-80. [3]. OTSSP167 Abrogates Mitotic Checkpoint through Inhibiting Multiple Mitotic Kinases. PLoS One. 2016 Apr 15;11(4):e0153518. [4]. The crystal structure of MPK38 in complex with OTSSP167, an orally administrative MELK selective inhibitor. Biochem Biophys Res Commun. 2014 Apr 25;447(1):7-11. |
| Additional Infomation |
We previously reported MELK (maternal embryonic leucine zipper kinase) as a novel therapeutic target for breast cancer. MELK was also reported to be highly upregulated in multiple types of human cancer. It was implied to play indispensable roles in cancer cell survival and indicated its involvement in the maintenance of tumor-initiating cells. We conducted a high-throughput screening of a compound library followed by structure-activity relationship studies, and successfully obtained a highly potent MELK inhibitor OTSSP167 with IC₅₀ of 0.41 nM. OTSSP167 inhibited the phosphorylation of PSMA1 (proteasome subunit alpha type 1) and DBNL (drebrin-like), which we identified as novel MELK substrates and are important for stem-cell characteristics and invasiveness. The compound suppressed mammosphere formation of breast cancer cells and exhibited significant tumor growth suppression in xenograft studies using breast, lung, prostate, and pancreas cancer cell lines in mice by both intravenous and oral administration. This MELK inhibitor should be a promising compound possibly to suppress the growth of tumor-initiating cells and be applied for treatment of a wide range of human cancer. [1] Maternal embryonic leucine zipper kinase (MELK) is upregulated in a variety of human tumors, and is considered an attractive molecular target for cancer treatment. We characterized the expression of MELK in gastric cancer (GC) and measured the effects of reducing MELK mRNA levels and protein activity on GC growth. MELK was frequently overexpressed in primary GCs, and higher MELK levels correlated with worse clinical outcomes. Reducing MELK expression or inhibiting kinase activity resulted in growth inhibition, G2/M arrest, apoptosis and suppression of invasive capability of GC cells in vitro and in vivo. MELK knockdown led to alteration of epithelial mesenchymal transition (EMT)-associated proteins. Furthermore, targeting treatment with OTSSP167 in GC patient-derived xenograft (PDX) models had anticancer effects. Thus, MELK promotes cell growth and invasiveness by inhibiting apoptosis and promoting G2/M transition and EMT in GC. These results suggest that MELK may be a promising target for GC treatment.[2] OTSSP167 was recently characterized as a potent inhibitor for maternal embryonic leucine zipper kinase (MELK) and is currently tested in Phase I clinical trials for solid tumors that have not responded to other treatment. Here we report that OTSSP167 abrogates the mitotic checkpoint at concentrations used to inhibit MELK. The abrogation is not recapitulated by RNAi mediated silencing of MELK in cells. Although OTSSP167 indeed inhibits MELK, it exhibits off-target activity against Aurora B kinase in vitro and in cells. Furthermore, OTSSP167 inhibits BUB1 and Haspin kinases, reducing phosphorylation at histones H2AT120 and H3T3 and causing mislocalization of Aurora B and associated chromosomal passenger complex from the centromere/kinetochore. The results suggest that OTSSP167 may have additional mechanisms of action for cancer cell killing and caution the use of OTSSP167 as a MELK specific kinase inhibitor in biochemical and cellular assays.[3] Murine protein serine/threonine kinase 38 (MPK38), also known as maternal embryonic leucine zipper kinase (MELK), has been associated with various human cancers and plays an important role in the formation of cancer stem cells. OTSSP167, a MELK selective inhibitor, exhibits a strong in vitro activity, conferring an IC50 of 0.41nM and in vivo effect on various human cancer xenograft models. Here, we report the crystal structure of MPK38 (T167E), an active mutant, in complex with OTSSP167 and describe its detailed protein-inhibitor interactions. Comparison with the previous determined structure of MELK bound to the nanomolar inhibitors shows that OTSSP167 effectively fits into the active site, thus offering an opportunity for structure-based development and optimization of MELK inhibitors.[4] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |