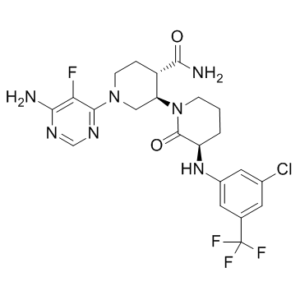
Vecabrutinib (formerly SNS-062; FP-182, BSK-4841 and BIIB-062) is a novel, potent, and reversible/noncovalent BTK and ITK inhibitor with Kd of 0.3 nM and 2.2 nM, respectively; Vecabrutinib shows an IC50 of 24 nM for ITK. SNS-062 does not require interaction with BTK C481 for inhibitory activity. SNS-062 has improved PK properties over ibrutinib, including greater bioavailability and a longer half-life. SNS-062 may offer an alternative treatment option to patients with acquired resistance to ibrutinib.
Physicochemical Properties
| Molecular Formula | C22H24CLF4N7O2 |
| Molecular Weight | 529.918276786804 |
| Exact Mass | 529.161 |
| CAS # | 1510829-06-7 |
| Related CAS # | 1270014-40-8 (SNS062-analog);1510829-06-7; 1947403-49-7; |
| PubChem CID | 72547837 |
| Appearance | White to light yellow solid powder |
| Density | 1.5±0.1 g/cm3 |
| Boiling Point | 776.0±60.0 °C at 760 mmHg |
| Flash Point | 423.1±32.9 °C |
| Vapour Pressure | 0.0±2.7 mmHg at 25°C |
| Index of Refraction | 1.621 |
| LogP | 3.13 |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 11 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 36 |
| Complexity | 809 |
| Defined Atom Stereocenter Count | 3 |
| SMILES | C1C[C@H](C(=O)N(C1)[C@H]2CN(CC[C@@H]2C(=O)N)C3=NC=NC(=C3F)N)NC4=CC(=CC(=C4)C(F)(F)F)Cl |
| InChi Key | QLRRJMOBVVGXEJ-XHSDSOJGSA-N |
| InChi Code | InChI=1S/C22H24ClF4N7O2/c23-12-6-11(22(25,26)27)7-13(8-12)32-15-2-1-4-34(21(15)36)16-9-33(5-3-14(16)19(29)35)20-17(24)18(28)30-10-31-20/h6-8,10,14-16,32H,1-5,9H2,(H2,29,35)(H2,28,30,31)/t14-,15+,16-/m0/s1 |
| Chemical Name | (3R,3'R,4'S)-1'-(6-amino-5-fluoropyrimidin-4-yl)-3-[3-chloro-5-(trifluoromethyl)anilino]-2-oxo[1,3'-bipiperidine]-4'-carboxamide |
| Synonyms | SNS-062; SNS 062; SNS062; BSK-4841; BSK4841; BSK 4841; FP182; FP 182; FP-182; BIIB-062; BIIB062; BIIB 062 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| ln Vitro | In human whole blood, vecabrutinib inhibits pBTK with a mean IC50 of 50 nM. Similar IC50 values (pBTK IC50: WT BTK 2.9 nM, C481S BTK 4.4 nM) are observed for vecabrutinib's inhibition of WT and C481S BTK [1]. Vecabrutinib's IC50 against WT BTK and C481S BTK in recombinant kinase assays was 4.6 nM and 1.1 nM, respectively. Vecabrutinib continues to be active against BTK mutant forms. Vecabrutinib demonstrated dose-dependent suppression of BTK similar to PCI-32765 in primary patient CLL cells via BTK phosphorylation immunoblotting. Vecabrutinib was six times more effective than PCI-32765 and more than 640 times more potent than acalabrutinib against C481S BTK. Vecabrutinib decreases primary CLL cell survival by 5.5% when HS5 stromal protection is present [2]. |
| ln Vivo | In rats and dogs, vecabrutinib exhibits a good oral bioavailability (%F > 40%) with a terminal half-life of five to six hours. At doses and durations much beyond PCI-32765, vecabrutinib was well tolerated [1]. |
| References |
[1]. Abstract C186: SNS-062 is a potent noncovalent BTK inhibitor with comparable activity against wild type BTK and BTK with an acquired resistance mutation. Molecular Cancer Therapeutics. December 2015 Volume 14, Issue 12 Supplement. [2]. Abstract 1207: SNS-062 demonstrates efficacy in chronic lymphocytic leukemia in vitro and inhibits C481S mutated Bruton tyrosine kinase. Cancer Research July 2017 Volume 77, Issue 13 Supplement. |
| Additional Infomation |
Vecabrutinib is under investigation in clinical trial NCT03037645 (Safety, PK, PD, and Antitumor Activity of Vecabrutinib (SNS-062) in B Lymphoid Cancers). Vecabrutinib is an orally available second-generation, reversible inhibitor of Bruton's tyrosine kinase (BTK; Bruton agammaglobulinemia tyrosine kinase), with potential antineoplastic activity. Upon administration, vecabrutinib non-covalently binds to and inhibits the activity of both wild-type and the C481S mutated form of BTK, a resistance mutation in the BTK active site in which cysteine is substituted for serine at residue 481. This prevents the activation of the B-cell antigen receptor (BCR) signaling pathway and BTK-mediated activation of downstream survival pathways. This leads to an inhibition of the growth of malignant B-cells that overexpress BTK. Compared to other BTK inhibitors, SNS-062 does not require interaction with the BTK C481 site and inhibits the proliferation of cells harboring the BTK C481S mutation. Other irreversible BTK inhibitors covalently bind to the C481 site to inhibit BTK's activity; the C481S mutation prevents that binding. BTK, a member of the Src-related BTK/Tec family of cytoplasmic tyrosine kinases, is overexpressed in B-cell malignancies; it plays an important role in the development, activation, signaling, proliferation and survival of B-lymphocytes. |
Solubility Data
| Solubility (In Vitro) | DMSO : ~125 mg/mL (~235.88 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (4.72 mM) (saturation unknown) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (4.72 mM) (saturation unknown) in 5% DMSO + 95% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (3.93 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 4: ≥ 2.08 mg/mL (3.93 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 5: ≥ 2.08 mg/mL (3.93 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.8871 mL | 9.4354 mL | 18.8708 mL | |
| 5 mM | 0.3774 mL | 1.8871 mL | 3.7742 mL | |
| 10 mM | 0.1887 mL | 0.9435 mL | 1.8871 mL |