Physicochemical Properties
| Molecular Formula | C13H16N5NA3O9P2S |
| Molecular Weight | 549.28 |
| Exact Mass | 548.9837051 |
| Related CAS # | MRS2365;436847-09-5 |
| PubChem CID | 168007094 |
| Appearance | Colorless to light yellow liquid |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 14 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 33 |
| Complexity | 792 |
| Defined Atom Stereocenter Count | 5 |
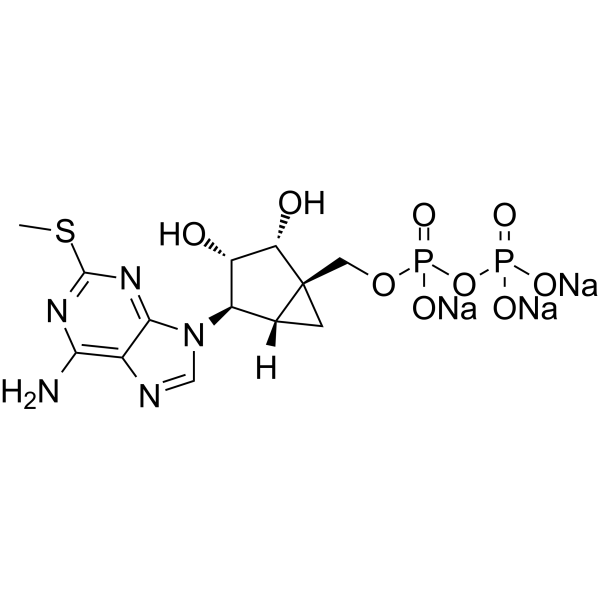
| SMILES | CSC1=NC(=C2C(=N1)N(C=N2)[C@@H]3[C@H]4C[C@]4([C@H]([C@H]3O)O)COP(=O)([O-])OP(=O)([O-])[O-])N.[Na+].[Na+].[Na+] |
| InChi Key | ZYPJOXVKTCEBCA-PGPREJRXSA-K |
| InChi Code | InChI=1S/C13H19N5O9P2S.3Na/c1-30-12-16-10(14)6-11(17-12)18(4-15-6)7-5-2-13(5,9(20)8(7)19)3-26-29(24,25)27-28(21,22)23;;;/h4-5,7-9,19-20H,2-3H2,1H3,(H,24,25)(H2,14,16,17)(H2,21,22,23);;;/q;3*+1/p-3/t5-,7-,8+,9+,13+;;;/m1.../s1 |
| Chemical Name | trisodium;[[(1R,2R,3S,4R,5S)-4-(6-amino-2-methylsulfanylpurin-9-yl)-2,3-dihydroxy-1-bicyclo[3.1.0]hexanyl]methoxy-oxidophosphoryl] phosphate |
| Synonyms | MRS2365; MRS2365 (trisodium); GLXC-01753; HY-108656A; [[(1R,2R,3S,4R,5S)-4-[6-Amino-2-(methylthio)-9H-purin-9-yl]-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl] diphosphoric acid mono ester trisodium salt |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | P2Y1 Receptor 0.4 nM (EC50) |
| ln Vitro |
Adenosine diphosphate (ADP)-induced platelet aggregation is attenuated by MRS2365 trisodium (1 µM or 3 µM; 2 min) [2]. ADP is the cognate agonist of the P2Y1, P2Y12, and P2Y13 receptors. With the goal of identifying a high potency agonist that selectively activates the P2Y1 receptor, Researchers examined the pharmacological selectivity of the conformationally constrained non-nucleotide analog (N)-methanocarba-2MeSADP (MRS2365) [(1′S,2′R, 3′S,4′R,5′S)-4-[(6-amino-2-methylthio-9H-purin-9-yl)-1-diphosphoryloxymethyl]bicyclo[3.1.0]hexane-2,3-diol] among the three ADP-activated receptors. Each P2Y receptor was expressed transiently in COS-7 cells, and inositol lipid hydrolysis was quantified as a measure of receptor activity. In the case of the Gi-linked P2Y12 and P2Y13 receptors, a chimeric G protein, Gαq/i, was coexpressed to confer a capacity of these Gi-linked receptors to activate phospholipase C. 2MeSADP (2-methylthio-ADP) was a potent agonist at all three receptors exhibiting EC50 values in the sub to low nanomolar range. In contrast, whereas (N)-methanocarba-2MeSADP was an extremely potent (EC50 = 1.2 ± 0.2 nM) agonist at the P2Y1 receptor, this non-nucleotide analog exhibited no agonist activity at the P2Y12 receptor and very low activity at the P2Y13 receptor. (N)-Methanocarba-2MeSADP also failed to block the action of 2MeSADP at the P2Y12 and P2Y13 receptors, indicating that the (N)-methanocarba analog is not an antagonist at these receptors. The P2Y1 receptor selectivity of (N)-methanocarba-2MeSADP was confirmed in human platelets where it induced the shape change promoted by P2Y1 receptor activation without inducing the sustained platelet aggregation that requires simultaneous activation of the P2Y12 receptor. These results provide the first demonstration of a high-affinity agonist that discriminates among the three ADP-activated P2Y receptors, and therefore, introduce a potentially important new pharmacological tool for delineation of the relative biological action of these three signaling proteins.[1] Adenosine diphosphate (ADP) initiates and maintains sustained aggregation of platelets through simultaneous activation of both the Gq-coupled P2Y1 receptor and the Gi-coupled P2Y12 receptor. Researchers recently described the synthesis and P2Y1 receptor-specific agonist activity of (N)-methanocarba-2MeSADP (MRS2365). Consequences of selective activation of the P2Y1 receptor by MRS2365 have been further examined in human platelets. Whereas MRS2365 alone only induced shape change, addition of MRS2365 following epinephrine treatment, which activates the Gi/z-linked, alpha2A-adrenergic receptor, resulted in sustained aggregation that was indistinguishable from that observed with ADP. Conversely, the platelet shape change promoted by ADP in the presence of the GPIIb/IIIa antagonist eptifibatide was similar to that promoted by MRS2365. Preaddition of the high affinity P2Y1 receptor antagonist MRS2500 inhibited the effect of MRS2365, whereas addition of MRS2500 subsequent to MRS2365 reversed the MRS2365-induced shape change. Preactivation of the P2Y1 receptor with MRS2365 for 2 min resulted in marked loss of capacity of ADP to induce aggregation as evidenced by a greater than 20-fold rightward shift in the concentration effect curve of ADP. This inhibitory effect of P2Y1 receptor activation was dependent on the concentration of MRS2365 (EC50 = 34 nm). The inhibitory effect of preincubation with MRS2365 was circumvented by activation of the Gq-coupled 5-HT2A receptor suggesting that MRS2365 induces loss of the ADP response as a consequence of desensitization of the Gq-coupled P2Y1 receptor. The time course of MRS2365-induced loss of aggregation response to epinephrine was similar to that observed with ADP. These results further demonstrate the P2Y1 receptor selectivity of MRS2365 and illustrate the occurrence of agonist-induced desensitization of the P2Y1 receptor of human platelets studied in the absence of P2Y12 receptor activation[2]. |
| ln Vivo |
In a dose-dependent way, MRS2365 trisodium (0.03-0.3 mg/kg; i.p.; single dose) considerably lessens mechanical allodynia in the male wistar rat neuropathy (Seltzer) model [3]. In a male wistar rat model of neuropathic pain, paw withdrawal threshold (PWT) is increased by MRS2365 trisodium (0.1-2 mg/kg; i.p.; single dose) [3]. In the wistar nerve damage (Seltzer) model, MRS2365 (0.03-0.3 mg/kg; intraperitoneal injection; single dose) substantially reduces mechanical allodynia in a coordinated dosage manner [3]. MRS2365, MRS2395 and UTP, but not the other compounds, significantly alleviated mechanical allodynia in the neuropathic pain model, with the following rank order of minimal effective dose (mED) values: MRS2365 > MRS2395 > UTP. All compounds had a dose-dependent analgesic action in acute pain except BBG, which elicited hyperalgesia at a single dose. The rank order of mED values in acute pain was the following: MRS2365 > MRS2395 > NF449 > NF023 > UDP = UTP > PPADS. MRS2365 and MRS2395 had a profound, while BBG had a mild effect on inflammatory pain, with a following rank order of mED values: MRS2395 > MRS2365 > BBG. None of the tested compounds had significant action on oedema evoked by intraplantar injection of CFA. Conclusions and implications: Our results show that antagonism at P2X1, P2Y12, and P2X7 receptors and agonism at P2Y1 receptors define promising therapeutic strategies in acute, neuropathic, and inflammatory pain respectively[3]. |
| Enzyme Assay |
IP-1 Assay. [4] IP-1, which is a metabolite of inositol 1,4,5-trisphosphate and downstream of Gq signaling, was detected by the IP-One Tb HTRF Kit, as described previously (Violin et al., 2010; Rajagopal et al., 2011). Briefly, after overnight growth, cells were pretreated with an antagonist for 20 minutes before the addition of agonist and incubated for another 60 minutes. Detection reagents were added as instructed by the manual from the manufacturer. The assay plates were read on a Mithras LB940 Reader or a PerkinElmer EnSpire plate reader using a time-resolved fluorescence ratio (665:620 nm). ERK1/2 Stimulation. [4] The method used was essentially as previously described (Gao et al., 2011, 2014). 1321N1 astrocytoma cells or U2OS cells expressing the human P2Y1R (30,000 cells/100 μl) were seeded in a 96-well plate in complete growth medium. After cell attachment, medium was removed and cells were serum starved overnight in 90 μl of serum-free medium. For Gαq-mediated ERK1/2 stimulation, cells were stimulated with agonist for 5 minutes. For β-arrestin2–mediated ERK1/2 stimulation, cells were incubated with agonist for 30 minutes after a pretreatment with the broad-spectrum protein kinase C (PKC) inhibitor GO6983 (10 µM) for 20 minutes (Lefkowitz and Shenoy, 2005; Hoffmann et al., 2008; Reiner et al., 2009). In both cases, cells were pretreated with an antagonist 20 minutes before the addition of agonist. After agonist treatment, the medium was removed and cells were lysed with 1× lysis buffer (20 μl) [AlphaScreen SureFire p-ERK1/2 (Thr202/Tyr204) Assay Kit; PerkinElmer]. Lysate (4 μl/well) was transferred to a 384-well ProxiPlate Plus (PerkinElmer). Acceptor beads were diluted 1:50 in a 1:5 mixture of activation buffer in reaction mix and added to the 384-well plate (5 μl/well). The plate was sealed and incubated for 2 hours at room temperature. Donor beads (2 μl) diluted 1:20 in dilution buffer were added, and the plate was incubated for another 2 hours at room temperature. The plate was measured using an EnVision multilabel reader using standard AlphaScreen settings. Lipofectamine 2000 was used for the transfection of β-arrestin2 siRNA (final concentration, 100 nM) according to the manufacturer manual. [35S]GTPγS Binding Assay. [4] The preparation of membranes from U2OS cells expressing human P2Y1R was as previously described (Gao et al., 2011). [35S]GTPγS binding was carried out in duplicate or triplicate by incubation for 30 minutes at 25°C in 200 μl of buffer containing 50 mM Tris HCl (pH 7.4), 1 mM EDTA, 1 mM MgCl2, 1 μM GDP, 1 mM dithiothreitol, 100 mM NaCl, 0.2 nM [35S]GTPγS, 0.5% bovine serum albumin, test agonists, and membrane suspension (10 μg protein/tube). Antagonists were added 20 minutes before the addition of agonists. The reaction was stopped by rapid filtration through Whatman GF/B filters, presoaked in 50 mM Tris HCl and 5 mM MgCl2 (pH 7.4). The filters were washed twice with 3 ml of the same buffer, and the retained radioactivity was measured using liquid scintillation counting. Nonspecific binding of [35S]GTPγS was measured in the presence of 10 μM unlabeled GTPγS. Label-Free DMR Measurement. [4] Label-free DMR measurement was performed on a PerkinElmer EnSpire multimode plate reader based on the EPIC optical biosensor technology using resonance waveguides. For the measurements, 50 μl of P2Y1R-expressing U2OS cells (2 × 104 cells/each well) were seeded into 96-well EnSpire cell assay microplates. The microplates were incubated in a humidified atmosphere containing 5% CO2 at 37°C overnight. Immediately before the experiments, the cells were washed three times with assay buffer (Hank’s balanced salt solution buffer containing 20 mM HEPES) and were allowed to equilibrate in the EnSpire multimode reader for 1 hour. After measuring baseline data, 10 μl of the test compounds were added and the cellular response was recorded continuously for 60 minutes. β-Arrestin2 Recruitment Assays. [4] The β-arrestin2 recruitment to the P2Y1R was assessed by DiscoverX PathHunter β-arrestin assay as described previously (Gao and Jacobson, 2008; Gao et al., 2014). In this assay, the GPCR is fused in frame with the small enzyme fragment ProLink and coexpressed in U2OS cells stably expressing a fusion protein of β-arrestin2 and the larger N-terminal deletion mutant of β-galactosidase (enzyme acceptor). GPCR activation stimulates binding of β-arrestin2 to the ProLink-tagged GPCR and forces complementation of the two enzyme fragments, resulting in the formation of an active β-galactosidase enzyme. This interaction leads to an increase in enzyme activity that can be measured using chemiluminescent PathHunter Detection Reagents (DiscoverX). For the measurement of P2Y1R-mediated recruitment, PathHunter U2OS cells expressing the human P2Y1R were grown in 96-well plates for 24 hours in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 μmol/ml glutamine. Cells were first treated with antagonists for 20 minutes and then treated with agonists for 60 minutes before adding detection reagents (mixture of 1 part Galacton Star substrate with 5 parts Emerald II Solution and 19 parts of PathHunter Cell Assay Buffer), and incubated at room temperature for 60 minutes before luminescence was measured. P2Y1R Internalization. [4] P2Y1R internalization was assessed using a PathHunter eXpress Activated GPCR Internalization Assay as instructed by the manufacturer. In this assay, PathHunter U2OS cells are engineered to coexpress an untagged GPCR, enzyme acceptor–tagged β-arrestin2, and a ProLink tag localized to the endosomes. Activation of the untagged GPCR induces β-arrestin recruitment, followed by internalization of the receptor/arrestin-enzyme acceptor complex in protein kinase-tagged endosomes, which forces complementation of the two β-galactosidase enzyme fragments, forming a functional enzyme that hydrolyzes substrate to generate a chemiluminescent signal. The method used for this assay was similar to that described in the β-arrestin2 recruitment assay, except that the agonist incubation time was 180 minutes. |
| Cell Assay |
Transfection of COS-7 Cells and Quantification of P2Y Receptor-Stimulated Phospholipase C Activity [1] COS-7 cells were seeded in 12-well culture dishes at a density of approximately 60,000 cells per well and maintained in high glucose Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in an atmosphere of 90% air/10% CO2. The indicated DNA vectors (in pcDNA3.1) were transfected using Fugene 6 transfection reagent (3 μl of Fugene/1 μg of DNA) according to the manufacturer’s protocol. The human P2Y12 and P2Y13 receptor DNA vectors (200 ng/well) were cotransfected with a DNA vector for Gαq/i (100 ng/well). Gαq/i is a chimeric construct of Gα subunits that confers to Gi-linked receptors a capacity to activate Gq and phospholipase C-β, and therefore, to promote inositol lipid hydrolysis in response to agonist-promoted receptor activation (Conklin et al., 1992). An expression vector for the human P2Y1 receptor, which couples to Gq and phospholipase C-β (Schachter et al., 1996; Waldo and Harden, 2004), was transfected (200 ng/well) in COS-7 cells in the absence of Gαq/i. Approximately 24 h after transfection, the culture medium was changed to inositol-free Dulbecco’s modified Eagle’s medium containing 1 μCi/well myo-[2-3H]inositol, and metabolic labeling proceeded for 12 to 16 h. Agonist-promoted accumulation of [3H]inositol phosphates was quantified subsequent to addition of 10 mM LiCl to inhibit inositol phosphate phosphatases. The reaction was stopped after 60 min by aspiration of the medium and the addition of 50 mM formic acid followed by neutralization with 150 mM NH4OH. [3H]Inositol phosphates were quantified by Dowex chromatography as previously described (Brown et al., 1991). Preparation and Assay of Washed Human Platelets [1] Blood was collected from healthy volunteers into syringes containing one-sixth final blood volume of anticoagulant (65 mM citric acid, 85 mM sodium citrate, and 110 mM dextrose). The blood was centrifuged at 180g for 15 min, and the supernatant (platelet rich plasma) was removed. The platelet rich plasma was centrifuged, and the platelets were resuspended in a buffer consisting of 137 mM NaCl, 2.7 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 3 mM NaH2PO4, 5 mM glucose, 10 mM HEPES (pH 7.4), and 0.2% bovine serum albumin. These centrifugations and washes were repeated twice followed by a final resuspension in the medium described above containing 0.25 U/ml apyrase. Platelet aggregation was measured using the optical mode of a Chrono-Log aggregometer. Four hundred and fifty microliters of platelet suspension containing 1 mg/ml fibrinogen were warmed to 37°C and stirred at 1000 rpm. Indicated concentrations of drugs were added to the sample, and aggregation was monitored for 8 min. Two modifications were included to obtain accurate estimations of platelet shape change. The offset mode of the Aggro/Link computer interface was applied, and the reference sample cuvette of the aggregometer included a platelet suspension equivalent to 50% of that present in cuvettes for drug testing. Scanning electron microscopy [2] Washed platelets, incubated with drugs as indicated, were fixed by addition of 50 μL of platelet suspension to 500 μL 4% glutaraldehyde in 0.1 m sodium cacodylate [cacodylic acid sodium salt – Na(CH3)2AsO23H2O) pH 7.2] followed by rotation for 30 min at room temperature. One hundred microliters of the fixed platelet suspension was spread onto poly-l-lysine-coated glass coverslips and incubated for 30 min. Following adsorption, the coverslips were rinsed once in 0.1 m sodium cacodylate buffer and dehydrated through a graded series of five ethanol washes (30%, 50%, 75%, 100%, and 100%). The coverslips were transferred in 100% ethanol to a critical point dryer (Balzers CPD-020; Bal-Tec AG, Balzers, Liechtenstein) and dried using carbon dioxide as the transition solvent. The coverslips were mounted on aluminum specimen supports with carbon adhesive tabs and coated with a 15 nm thickness of gold–palladium metal (60:40 alloy) using a Hummer × sputter coater. Samples were examined with a Cambridge Stereoscan 200 scanning electron microscope operating at an acceleration voltage of 20 kV, 20 mm working distance, and 40° tilt. |
| Animal Protocol |
Animal/Disease Models: Male Wistar rats with neuropathic pain (250-350 g)[3]. Doses: 0.03, 0.1, 0.3, 1 and 2 mg/kg. Route of Administration: intraperitoneal (ip)injection; single dose. Experimental Results: Relieved mechanical allodynia and increased the paw withdrawal threshold. Background and purpose: This study was undertaken to compare the analgesic activity of antagonists acting at P2X1, P2X7, and P2Y12 receptors and agonists acting at P2Y1, P2Y2, P2Y4, and P2Y6 receptors in neuropathic, acute, and inflammatory pain. Experimental approach: The effect of the wide spectrum P2 receptor antagonist PPADS, the selective P2X7 receptor antagonist Brilliant Blue G (BBG), the P2X1 receptor antagonist (4,4',4'',4-[carbonylbis(imino-5,1,3-benzenetriyl-bis(carbonylimino))]tetrakis-1,3-benzenedisulfonic acid, octasodium salt (NF449) and (8,8'-[carbonylbis(imino-3,1-phenylenecarbonylimino)]bis-1,3,5-naphthalene-trisulphonic acid, hexasodium salt (NF023), the P2Y12 receptor antagonist (2,2-dimethyl-propionic acid 3-(2-chloro-6-methylaminopurin-9-yl)-2-(2,2-dimethyl-propionyloxymethyl)-propylester (MRS2395), the selective P2Y1 receptor agonist ([[(1R,2R,3S,4R,5S)-4-[6-amino-2-(methylthio)-9H-purin-9-yl]-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl] diphosphoric acid mono ester trisodium salt (MRS2365), the P2Y2/P2Y4 agonist uridine-5'-triphosphate (UTP), and the P2Y4/P2Y6 agonist uridine-5'-diphosphate (UDP) were examined on mechanical allodynia in the Seltzer model of neuropathic pain, on acute thermal nociception, and on the inflammatory pain and oedema induced by complete Freund's adjuvant (CFA).[3] |
| References |
[1]. Induction of novel agonist selectivity for the ADP-activated P2Y1 receptor versus the ADP-activated P2Y12 and P2Y13 receptors by conformational constraint of an ADP analog. J Pharmacol Exp Ther. 2004 Dec;311(3):1038-43. [2]. (N)-methanocarba-2MeSADP (MRS2365) is a subtype-specific agonist that induces rapid desensitization of the P2Y1 receptor of human platelets. J Thromb Haemost. 2006 Apr;4(4):861-8. [3]. A comparative analysis of the activity of ligands acting at P2X and P2Y receptor subtypes in models of neuropathic, acute and inflammatory pain. Br J Pharmacol. 2010 Mar;159(5):1106-17. [4]. Distinct Signaling Patterns of Allosteric Antagonism at the P2Y1 Receptor. Mol Pharmacol. 2017 Nov;92(5):613-626. |
| Additional Infomation |
Traditionally, G protein-coupled receptor antagonists are classified as competitive or noncompetitive and surmountable or insurmountable based on functional antagonism. P2Y1 receptor (P2Y1R) structures showed two antagonists binding to two spatially distinct sites: nucleotide MRS2500 (orthosteric, contacting the helical bundle) and urea BPTU (allosteric, on the external receptor surface). However, the nature of their P2Y1R antagonism has not been characterized. Here we characterized BPTU antagonism at various signaling pathways activated by structurally diverse agonists. BPTU rightward shifted the concentration-response curves of both 2-methylthioadenosine 5'-diphosphate trisodium salt and MRS2365 (5'-diphosphates) in some signaling events, such as extracellular signal-regulated kinase 1/2 and label free, in a parallel manner without affecting the maximum agonist effect (Emax) but antagonized insurmountably (suppressed agonist Emax) in signaling events such as guanosine 5'-3-O-(thio)triphosphate binding and β-arrestin2 recruitment. However, with dinucleotide Ap4A as an agonist, BPTU suppressed the Emax insurmountably in all signaling pathways. By comparison, MRS2500 behaved as surmountable antagonist rightward-shifting concentration-response curves of all three agonists in a parallel manner for all signaling pathways measured. Thus, we demonstrated a previously undocumented phenomenon that P2Y1R antagonism patterns could vary in different signaling pathways, which could be related to conformational selection, signaling amplification, and probe dependence. This phenomenon may apply generally to other receptors considering that antagonism by a specific ligand is often not compared at multiple signaling pathways. Thus, antagonism can be surmountable or insurmountable depending on the signaling pathways measured and the agonists used, which should be of broad relevance to drug discovery and disease treatment.[4]
In the case of (N)-methanocarba-2MeSADP, we have illustrated that ring constrained (N)-methanocarba substitution combined with another functionality increases both agonist potency and, as illustrated here, receptor selectivity among ADP-activated P2Y receptors. Combined with our previous results illustrating that (N)-methanocarba-2MeSADP does not activate the Gq/phospholipase C-coupled P2Y2, P2Y4, P2Y6, and P2Y11 receptors (Ravi et al., 2002), the current results have significant implications for development of agonists of high potency and selectivity for other subfamilies of receptors within the P2Y receptor family. [1] In summary, the studies outlined above have demonstrated that MRS2365 activates the P2Y1 receptor with high potency and selectivity as observed by hallmark features (i.e. shape change/pseudopod formation), leads to rapid P2Y1 receptor desensitization with no observable action at the P2Y12 receptor, and attenuates ADP-induced aggregation upon subsequent agonist challenge. MRS2365 is a novel pharmacological tool that should prove highly useful in discrete analysis of the bifurcating ADP-induced signaling pathways in platelets.[2] Thus, a principal novel finding of our study is the potent analgesic action of the selective P2Y1 receptor agonist MRS2365 in neuropathic and acute pain. In addition, although it exhibited a weaker potency, MRS2365 also alleviated inflammatory pain. Although the anti-nociceptive effect of agonists of P2Y1 receptors such as ADP and ADP-γ-S have been already demonstrated in acute and neuropathic pain (Okada et al., 2002; Gerevich et al., 2004), none of these compounds display selectivity towards the P2Y1 receptor. For instance, ADP binds preferentially to P2Y12 and P2Y13 receptors as well (Abbracchio et al., 2006). MRS2365 acts in the nanomolar range and selectively activates recombinant P2Y1 receptors but not P2Y12 and P2Y13 receptors in vitro (Chhatriwala et al., 2004), which renders likely that its potent in vivo actions are mediated by P2Y1 receptors, although we cannot entirely rule out an additional action on other receptors. [3] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.8206 mL | 9.1028 mL | 18.2057 mL | |
| 5 mM | 0.3641 mL | 1.8206 mL | 3.6411 mL | |
| 10 mM | 0.1821 mL | 0.9103 mL | 1.8206 mL |