JI-101 is an orally bioactive multi-kinase inhibitor of vascular endothelial growth factor receptor 2 (VEGFR2), platelet-derived growth factor receptor beta (PDGFRb), and the ephrin B4 receptor B4 (EphB4) with potential antiangiogenic and antineoplastic activities. JI-101 has demonstrated strong anticancer activity against a range of cancer cell lines and xenografts both in vitro and in vivo. For the treatment of solid tumors, it is presently in Phase II clinical development. In all preclinical and human liver microsomes, JI-101 is found to be stable. Across the tested species of liver microsomes, the percentage metabolized ranges from 3.03 to 3.95. Rat, dog, and human liver microsomes are the next in order of percentage metabolized, which is comparatively higher in mice liver microsomes.
Physicochemical Properties
| Molecular Formula | C₂₂H₂₀BRN₅O₂ | |
| Molecular Weight | 466.33 | |
| Exact Mass | 465.08 | |
| Elemental Analysis | C, 56.66; H, 4.32; Br, 17.13; N, 15.02; O, 6.86 | |
| CAS # | 900573-88-8 | |
| Related CAS # |
|
|
| PubChem CID | 11691242 | |
| Appearance | White to off-white solid powder | |
| LogP | 5.098 | |
| Hydrogen Bond Donor Count | 3 | |
| Hydrogen Bond Acceptor Count | 4 | |
| Rotatable Bond Count | 5 | |
| Heavy Atom Count | 30 | |
| Complexity | 582 | |
| Defined Atom Stereocenter Count | 0 | |
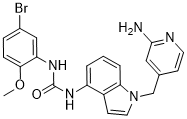
| SMILES | O=C(NC1C(OC)=CC=C(Br)C=1)NC1C2C=CN(C=2C=CC=1)CC1C=C(N)N=CC=1 |
|
| InChi Key | ZXBFYBLSJMEBEP-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C22H20BrN5O2/c1-30-20-6-5-15(23)12-18(20)27-22(29)26-17-3-2-4-19-16(17)8-10-28(19)13-14-7-9-25-21(24)11-14/h2-12H,13H2,1H3,(H2,24,25)(H2,26,27,29) | |
| Chemical Name | 1-[1-[(2-aminopyridin-4-yl)methyl]indol-4-yl]-3-(5-bromo-2-methoxyphenyl)urea | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
VEGFR2; PDGFRβ
Triple kinase inhibitor [1] |
| ln Vitro |
JI-101 is discovered to be stable in human liver microsomes and all preclinical models. The percentage of metabolized liver microsomes in the tested species ranges from 3.03 to 3.95. Mice liver microsomes have a comparatively higher percentage of metabolized material than do dog, human, and rat liver microsomes[1]. |
| ln Vivo | JI-101 and its mono- and di-hydroxy metabolites are eliminated through bile. After being taken orally, JI-101 is quickly absorbed and reaches its Cmax in two hours. The JI-101 t1/2 for the oral and intravenous routes are determined to be 2.66±0.13 h and 1.75±0.79 h, respectively. By intravenous route, the Cl and Vd for JI-101 are determined to be 13.0±2.62 mL/min/kg and 2.11±1.42 L/kg, respectively. JI-101 has a wide tissue distribution, with lung tissue absorbing it most quickly. JI-101 has an overall oral bioavailability of 55%, and its principal route of elimination is through feces[1]. |
| Enzyme Assay | JI-101 is an oral bioactive multi-kinase inhibitor that has the ability to inhibit the activities of platelet-derived growth factor receptor beta (PDGFRb), vascular endothelial growth factor receptor 2 (VEGFR2), and ephrin B4 receptor B4 (EphB4). It may also have antiangiogenic and antineoplastic properties. |
| Cell Assay | It has been discovered that JI-101 is stable in both human and preclinical liver microsomes. The percentage of metabolized liver microsomes in the tested species ranges from 3.03 to 3.95. Mice liver microsomes have a comparatively higher percentage of metabolized material compared to dog, human, and rat liver microsomes. |
| Animal Protocol |
Rats: In a preliminary parallel-group study, male S.D. rats are used to assess the pharmacokinetics and bioavailability of JI-101. JI-101 is administered intravenously (IV) via tail vein to four rats weighing between 195 and 210 g, orally by gavage, at a dose of 30 mg/kg. At pre-dose, 0.12 (i.v. only), 0.25, 0.5, 1, 2, 4, 8, 10 (oral only), and 24 hours, serial blood samples (100 μL) are taken from the retro-orbital plexus. Samples of blood are taken in tubes with K2 EDTA as the anticoagulant, centrifuged for 5 minutes at 4 °C to separate the plasma, and then frozen at -80±10 °C until analysis[1]. 1. Pharmacokinetic and tissue distribution study: Male Wistar rats (200-250 g) were randomly divided into two groups (n=6/group) for intravenous (i.v.) and oral (p.o.) administration. JI-101 was dissolved in a mixture of polyethylene glycol 400 (PEG400) and normal saline (50:50, v/v) for i.v. injection at a dose of 5 mg/kg, and suspended in 0.5% carboxymethylcellulose sodium (CMC-Na) for oral gavage at a dose of 20 mg/kg. Blood samples (0.3 mL) were collected from the retro-orbital plexus at 0.083, 0.25, 0.5, 1, 2, 4, 6, 8, 12, and 24 hours post-administration, anticoagulated with heparin, and centrifuged to separate plasma. At 1, 4, and 8 hours post-administration, rats were sacrificed, and tissues including heart, liver, spleen, lung, kidney, brain, muscle, fat, and gastrointestinal tract were collected, rinsed with normal saline, blotted dry, and weighed [1] 2. Metabolite identification study: Another group of male Wistar rats (200-250 g, n=3) received a single oral dose of 20 mg/kg JI-101 (suspended in 0.5% CMC-Na). Urine and feces were collected at 0-24, 24-48, and 48-72 hours post-administration. Plasma samples were collected at 1, 4, and 8 hours, and liver tissue was collected at 8 hours post-sacrifice. All samples were stored at -80℃ until metabolite analysis [1] |
| ADME/Pharmacokinetics |
1. Absorption: After oral administration of 20 mg/kg JI-101 in rats, the absolute bioavailability was 12.3%. The peak plasma concentration (Cmax) was 0.87 μg/mL, achieved at 1.5 hours (Tmax) [1] 2. Distribution: JI-101 showed extensive tissue distribution in rats. At 1 hour post-oral administration (20 mg/kg), the highest drug concentrations were detected in the liver (6.2 μg/g) and kidney (4.8 μg/g), followed by lung (2.5 μg/g), spleen (1.9 μg/g), heart (1.6 μg/g), gastrointestinal tract (1.5 μg/g), muscle (0.9 μg/g), and fat (0.7 μg/g). The brain concentration was 0.12 μg/g, indicating poor blood-brain barrier penetration. The volume of distribution (Vd) after intravenous administration (5 mg/kg) was 3.8 L/kg [1] 3. Metabolism: JI-101 was metabolized primarily in the liver. Four putative metabolites (M1-M4) were identified in plasma, urine, feces, and liver tissue using liquid chromatography-tandem mass spectrometry (LC-MS/MS). The main metabolic pathways included oxidation (M1, M2) and hydrolysis (M3, M4), with M1 (monohydroxylated metabolite) being the major metabolite [1] 4. Excretion: Within 72 hours post-oral administration, 68.2% of the administered dose was excreted in feces (mostly as unchanged drug and M1), and 12.5% was excreted in urine (primarily as metabolites M2-M4). The total excretion rate was 80.7% [1] 5. Elimination: After intravenous administration (5 mg/kg), the plasma elimination half-life (t1/2) was 6.8 hours, and the plasma clearance (CL) was 0.35 L/h/kg [1] |
| Toxicity/Toxicokinetics |
1. Plasma protein binding: JI-101 showed 94.6% plasma protein binding in rat plasma, determined by equilibrium dialysis [1] 2. Acute toxicity: No obvious toxic symptoms (e.g., weight loss, lethargy, abnormal behavior) were observed in rats after single oral administration of JI-101 at doses up to 20 mg/kg or intravenous administration at 5 mg/kg within 24 hours of observation [1] |
| References |
[1]. Pharmacokinetics, tissue distribution and identification of putative metabolites of JI-101 - a novel triple kinase inhibitor in rats. Arzneimittelforschung. 2012 Jan;62(1):27-34. |
| Additional Infomation |
JI-101 has been used in trials studying the treatment of Cancer, Colon Cancer, Neuroendocrine, Ovarian Cancer, and Advanced Solid Tumors. Angiogenesis Inhibitor JI-101 is an orally active inhibitor of vascular endothelial growth factor receptor 2 (VEGFR2), platelet-derived growth factor receptor beta (PDGFRb), and the ephrin B4 receptor B4 (EphB4) with potential antiangiogenic and antineoplastic activities. Angiogenesis inhibitor JI-101 binds to and inhibits VEGFR2, PDGFRb and EphB4, which may inhibit tumor angiogenesis and, so, cellular proliferation in tumor cells overexpressing VEGFR2, PDGFRb and EphB4. The receptor tyrosine kinases VEGFR2, PDGFRb and EphB4 may be overexpressed in a number of different cancer cell types and may play crucial roles in tumor angiogenesis. 1. JI-101 is a novel triple kinase inhibitor with potential antitumor activity (not evaluated in this literature). It exhibits poor oral bioavailability (12.3%) but extensive tissue distribution, with high concentrations in metabolic organs (liver, kidney) [1] 2. The drug is metabolized mainly via hepatic oxidation and hydrolysis, producing four putative metabolites, and is primarily excreted in feces. Its poor blood-brain barrier penetration suggests limited utility for central nervous system tumors [1] 3. JI-101 has high plasma protein binding (94.6), which may affect its distribution and potential drug-drug interactions (not further evaluated in the literature) [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.36 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.36 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.36 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1444 mL | 10.7220 mL | 21.4440 mL | |
| 5 mM | 0.4289 mL | 2.1444 mL | 4.2888 mL | |
| 10 mM | 0.2144 mL | 1.0722 mL | 2.1444 mL |