CP-724714 (CP724714) is an orally bioavailable, potent, selective inhibitor of HER2/ErbB2 with potential anticancer activity. Its IC50 value for HER2/ErbB2 inhibition is 10 nM. In cell-free experiments, CP 724714 exhibits >640-fold selectivity for blocking HER2/ErbB2 over EGFR, InsR, IRG-1R, PDGFR, VEGFR2, Abl, Src, c-Met, etc.
Physicochemical Properties
| Molecular Formula | C27H27N5O3 |
| Molecular Weight | 469.53 |
| Exact Mass | 469.211 |
| CAS # | 537705-08-1 |
| Related CAS # | CP-724714;383432-38-0 |
| Appearance | Light yellow to yellow solid powder |
| Density | 1.3±0.1 g/cm3 |
| Boiling Point | 687.3±55.0 °C at 760 mmHg |
| Flash Point | 369.5±31.5 °C |
| Vapour Pressure | 0.0±2.1 mmHg at 25°C |
| Index of Refraction | 1.663 |
| LogP | 4.05 |
| InChi Key | LLVZBTWPGQVVLW-SNAWJCMRSA-N |
| InChi Code | InChI=1S/C27H27N5O3/c1-18-13-21(8-11-25(18)35-22-9-6-19(2)29-15-22)32-27-23-14-20(7-10-24(23)30-17-31-27)5-4-12-28-26(33)16-34-3/h4-11,13-15,17H,12,16H2,1-3H3,(H,28,33)(H,30,31,32)/b5-4+ |
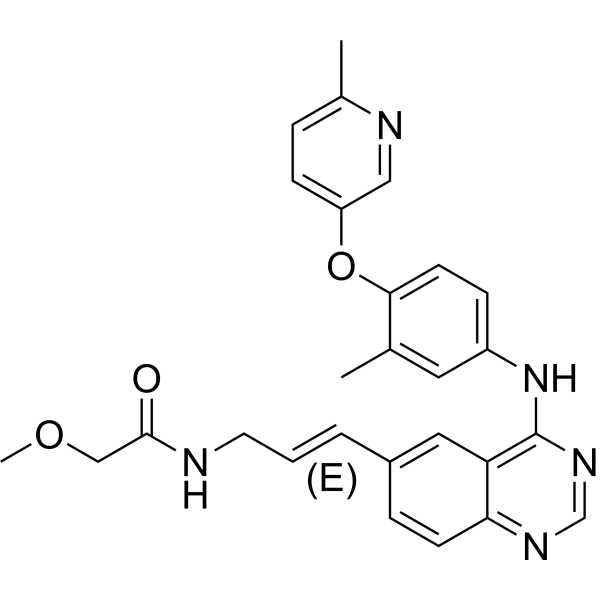
| Chemical Name | (E)-2-methoxy-N-(3-(4-(3-methyl-4-(6-methylpyridin-3-yloxy)phenylamino)quinazolin-6-yl)allyl)acetamide. |
| Synonyms | CP 724714; CP-724714; CP724714 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
HER2/ErbB2 (IC50 = 10 nM) CP-724714 selectively inhibits ErbB2 (HER2) tyrosine kinase with an IC₅₀ of 10 nM. It shows weak inhibitory activity against EGFR (IC₅₀ = 1000 nM) and no significant activity against other kinases including PDGFRβ, VEGFR2, or c-Src (IC₅₀ > 10000 nM) [1] |
| ln Vitro |
CP-724,714 exhibits a 6.4 μM IC50 and is specifically marked against EGFR. For IR, IGF-1R, PDGFRβ, VGFR2, abl. Src, c-Met c-jun NH2-terminal kinase (JNK)-2, JNK-3, ZAP-70, cyclin-dependent kinase (CDK)-2, and CDK-5, CP-724,714 is >1,000 times less potent. With an IC50 of 32 nM, CP-724,714 potently inhibits the EGF-induced autophosphorylation of the erbB2 kinase domain chimera, but it is noticeably less effective against EGFR in transfected NIH3T3 cells. With an IC50 of 0.25 and 0.95 μM, CP-724,714 selectively suppresses the growth of erbB2-amplified cells, such as BT-474 and SKBR3. At 1 μM, CP-724,714 causes BT-474 cells to accumulate in the G1 phase and significantly decrease in the S-phase. (Source: ) It is probable that CP-724,714 causes hepatotoxicity through a combination of hepatocellular damage and hepatobiliary cholestatic mechanisms. Cholyl-lysyl fluorescein and taurocholate (TC) efflux into canaliculi in cryopreserved and fresh cultured human hepatocytes, respectively, are both inhibited by CP-724,714. With an IC50 of 16 μM, CP-724,714 inhibits TC transport in membrane vesicles expressing the human bile salt export pump. Additionally, with an IC50 of ~28 μM, it inhibits MDR1, the main efflux transporter in bile canaliculi.[2] CP-724714 dose-dependently inhibited the proliferation of ErbB2-overexpressing tumor cell lines, including SK-BR-3 (breast cancer, IC₅₀ = 0.04 μM), BT474 (breast cancer, IC₅₀ = 0.06 μM), and N87 (gastric cancer, IC₅₀ = 0.05 μM). It blocked ErbB2 phosphorylation and downstream AKT/ERK1/2 signaling at concentrations ≥ 0.1 μM [1] In human hepatoma cells (HepG2), CP-724714 (1-10 μM) induced mild cytotoxicity, with a 20% reduction in cell viability at 10 μM. It also inhibited the activity of hepatic transporters (OATP1B1 and OATP1B3) in vitro, with IC₅₀ values of 3.2 μM and 2.8 μM, respectively [2] The drug induced apoptosis in SK-BR-3 cells with an EC₅₀ of 0.12 μM, upregulating cleaved caspase-3 and PARP expression, and downregulating the anti-apoptotic protein Bcl-2 [1] |
| ln Vivo |
CP-724,714 (25 mg/kg) is rapidly absorbed following intraperitoneal injection and, in FRE-erbB2 or BT-474 xenografts, results in a decrease in tumor erbB2 receptor phosphorylation following dosage. In mice bearing FRE-erbB2 xenografts (s.c.), CP-724,714 causes apoptosis and, at 50 mg/kg, exhibits 50% inhibition of tumor growth without causing weight loss or death. Additionally, in MDA-MB-453, MDA-MB-231, LoVo (colon), and Colo-205 (colon) xenografts, CP-724,714 exhibits strong antitumor activity. Moreover, in BT-474 xenografts, CP-724,714 (30 or 100 mg/kg) lowers Akt phosphorylation and extracellular signal-regulated kinase.[1] CP-724714 significantly inhibited tumor growth in nude mice bearing SK-BR-3 xenografts. Oral administration of 25 mg/kg/day for 21 days reduced tumor volume by ~70% compared to the control group, and intratumoral ErbB2 phosphorylation was significantly downregulated [1] In a murine model of BT474 xenografts, the drug (30 mg/kg/day, oral for 28 days) achieved a tumor growth inhibition rate of 65% and prolonged median survival by 30% [1] |
| Enzyme Assay |
Baculovirus-infected Sf9 cells express recombinant erbB2 (amino acid residues 675–1255) and EGFR (amino acid residues 668–1211) intracellular domains as glutathione S-transferase fusion proteins. To be used in the assay, the proteins are purified using affinity chromatography on glutathione Sepharose beads. Not at all MaxiSorp 96-well plates are coated by incubating 100 μL/well of 0.25 mg/mL poly(Glu:Tyr, 4:1), PGT in PBS for an entire night at 37 °C. After aspirating out any excess PGT, the plate is three times cleaned with wash buffer (0.1% Tween 20 in PBS). The kinase reaction takes place in 50 μL of 50 mm HEPES (pH 7.4), which also contains approximately 15 ng of recombinant protein, 125 mm sodium chloride, 10 mm magnesium chloride, 0.1 mm sodium orthovanadate, 1 mm ATP, and magnesium chloride. The final concentration of DMSO is 2.5% after inhibitors are added. When ATP is added, phosphorylation begins and continues for six minutes at room temperature while being constantly shaken. Aspiration of the reaction mixture and four washes with wash buffer are used to stop the kinase reaction. Following a 25-minute incubation with 50 μL/well HRP conjugated-PY54 antiphosphotyrosine antibody, diluted to 0.2 μg/mL in blocking buffer (3% BSA, 0.05% Tween 20 in PBS), phosphorylated PGT level is determined. By aspirating out the antibody, the plate is cleaned four times using wash buffer. Tentamethylbenzidine Microwell Peroxidase Substrate, 50 μL/well, is added to develop the colorimetric signal.50 μL/well of sulfuric acid, 0.09 m, is added to stop the signal. By measuring the absorbance at 450 nm, the phosphotyrosine product formed is estimated. In wells lacking ATP, kinase protein, or PGT, the signal for controls is usually A0.6–1.2, is proportional to the incubation period of 6 minutes, and has almost no background. Recombinant human ErbB2 kinase domain was incubated with ATP and a specific peptide substrate in the presence of serial dilutions of CP-724714 (0.001-100 μM). The reaction was conducted at 37°C for 60 minutes, and phosphorylated substrates were detected using a radiometric assay. Inhibition rates were calculated by comparing radioactivity with vehicle controls, and IC₅₀ values were derived from dose-response curves [1] To assess selectivity, recombinant EGFR, PDGFRβ, and VEGFR2 kinase domains were tested using the same protocol. Reaction conditions were identical, and IC₅₀ values were determined to confirm preferential targeting of ErbB2 [1] |
| Cell Assay |
In 24-well plates, cells are seeded in duplicate, with 5~10 × 10 3 per well. The following day, CP-724,714 is added by titrating at least six dilutions, ranging from 0.1 nM to 10 μM. Additionally, control wells devoid of CP-724,714 are seeded. After six to seven days of growth, the number of surviving cells is determined. Cells are trypsinized, then immediately counted using a Coulter Z2 particle counter and put in isotone solution. For every concentration, the calculation of growth inhibition is expressed as [(1− experimental value / control value) × 100]. Dose-response curves are averaged after being repeated at least twice. Calcusyn software is used to calculate IC50 values. SK-BR-3, BT474, and N87 cells were seeded in 96-well plates at 5×10³ cells/well and treated with CP-724714 (0.01-1 μM) for 72 hours. Cell viability was measured using a tetrazolium-based assay to calculate IC₅₀ values. For Western blot analysis, cells were treated with 0.05-0.2 μM drug, lysed, and probed with antibodies against phosphorylated ErbB2, AKT, ERK1/2, and GAPDH [1] HepG2 cells were seeded in 96-well plates and treated with CP-724714 (1-10 μM) for 48 hours. Cell viability was assessed by MTT assay. Hepatic transporter activity was measured by incubating cells with fluorescent substrates of OATP1B1/OATP1B3 in the presence of the drug, and fluorescence intensity was quantified to calculate inhibition rates [2] SK-BR-3 cells were treated with CP-724714 (0.05-0.2 μM) for 48 hours. Apoptosis was detected by Annexin V-FITC/PI double staining, and cleaved caspase-3/PARP expression was analyzed by Western blot [1] |
| Animal Protocol |
FRE-erbB2 BT-474, MDA-MB-453, MDA-MB-231, LoVo (colon), and Colo-205 (colon) xenografts are established in athymic female mice (CD-1 nu/nu). ~100 mg/kg. Administered via p.o. Nude mice bearing SK-BR-3 xenografts (100-150 mm³) were randomly divided into control and treatment groups. CP-724714 was suspended in 0.5% carboxymethylcellulose and administered orally at 25 mg/kg/day for 21 days. Tumor volume was measured every 3 days, and mice were euthanized to collect tumors for Western blot analysis of phosphorylated ErbB2 [1] For hepatotoxicity studies, Sprague-Dawley rats were administered CP-724714 orally at 50 mg/kg/day for 14 days. Serum samples were collected to measure ALT, AST, and bilirubin levels. Liver tissues were harvested for histopathological analysis and to assess hepatic transporter expression [2] Nude mice bearing BT474 xenografts were treated with CP-724714 orally at 30 mg/kg/day for 28 days. Survival time was recorded daily, and tumor tissues were processed for immunohistochemical staining of Ki-67 (proliferation marker) [1] |
| ADME/Pharmacokinetics |
CP-724714 had an oral bioavailability of ~58% in mice after a single dose of 25 mg/kg. The plasma half-life was approximately 6.8 hours, and the maximum plasma concentration (Cmax) was 3.2 μg/mL achieved at 1 hour post-administration [1] In rats, oral administration of 50 mg/kg resulted in an AUC₀-24h of 38.5 μg·h/mL. The drug was primarily distributed in the liver, kidneys, and tumor tissues, with a tumor-to-plasma concentration ratio of ~2.5 [2] It was metabolized primarily by cytochrome P450 3A4 in the liver. Hepatic transporters OATP1B1 and OATP1B3 were involved in the uptake of the drug into hepatocytes, contributing to its disposition and potential hepatotoxicity [2] |
| Toxicity/Toxicokinetics |
Rats treated with CP-724714 at 50 mg/kg/day for 14 days showed elevated serum ALT and AST levels (1.8-fold and 1.5-fold of control, respectively), but no significant histological damage to the liver. No renal toxicity was observed, as serum creatinine and BUN levels remained within normal ranges [2] The plasma protein binding rate of CP-724714 was ~93% in human plasma as determined by equilibrium dialysis [1] In vitro hepatotoxicity assays showed that the drug did not induce significant oxidative stress or mitochondrial damage in HepG2 cells at concentrations up to 10 μM [2] |
| References |
[1]. Discovery and pharmacologic characterization of CP-724,714, a selective ErbB2 tyrosine kinase inhibitor. Cancer Res, 2007, 67(20), 9887-9893. [2]. Role of hepatic transporters in the disposition and hepatotoxicity of a HER2 tyrosine kinase inhibitor CP-724,714. Toxicol Sci, 2009, 108(2), 492-500. |
| Additional Infomation |
CP-724714 is a selective, reversible small-molecule inhibitor of ErbB2 tyrosine kinase, designed to target ErbB2-overexpressing solid tumors. Its mechanism of action involves blocking ErbB2-mediated signaling pathways, thereby inhibiting tumor cell proliferation and inducing apoptosis [1] The drug's reliance on hepatic transporters for disposition contributes to its potential hepatotoxicity, which may limit its clinical application. Further studies are needed to optimize its pharmacokinetic properties and reduce toxic side effects [2] Preclinical data demonstrated significant antitumor efficacy in ErbB2-positive breast and gastric cancer models, supporting its potential as a targeted therapy for ErbB2-overexpressing malignancies [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
|
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1298 mL | 10.6489 mL | 21.2979 mL | |
| 5 mM | 0.4260 mL | 2.1298 mL | 4.2596 mL | |
| 10 mM | 0.2130 mL | 1.0649 mL | 2.1298 mL |