Physicochemical Properties
| Molecular Formula | C22H22F3N3O3 |
| Molecular Weight | 433.42 |
| Exact Mass | 433.161 |
| Elemental Analysis | C, 60.96; H, 5.12; F, 13.15; N, 9.69; O, 11.07 |
| CAS # | 1372540-25-4 |
| Related CAS # | 1372540-25-4;2108806-07-9 (HCl);1372540-91-4 (tris); |
| PubChem CID | 56949517 |
| Appearance | white solid powder |
| Density | 1.4±0.1 g/cm3 |
| Boiling Point | 641.3±55.0 °C at 760 mmHg |
| Flash Point | 341.7±31.5 °C |
| Vapour Pressure | 0.0±2.0 mmHg at 25°C |
| Index of Refraction | 1.606 |
| LogP | 3.61 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 8 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 31 |
| Complexity | 643 |
| Defined Atom Stereocenter Count | 0 |
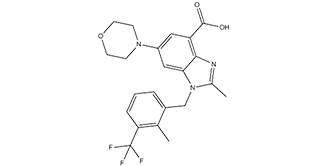
| SMILES | CC1=NC2=C(C(O)=O)C=C(C=C2N1CC3=CC=CC(C(F)(F)F)=C3C)N4CCOCC4 |
| InChi Key | XTKLTGBKIDQGQL-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C22H22F3N3O3/c1-13-15(4-3-5-18(13)22(23,24)25)12-28-14(2)26-20-17(21(29)30)10-16(11-19(20)28)27-6-8-31-9-7-27/h3-5,10-11H,6-9,12H2,1-2H3,(H,29,30) |
| Chemical Name | 2-methyl-1-(2-methyl-3-(trifluoromethyl)benzyl)-6-morpholino-1H-benzo[d]imidazole-4-carboxylic acid. |
| Synonyms | GSK 2636771; GSK2636771; GSK-2636771 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
PI3Kβ (Ki = 0.89 nM); PI3Kβ (IC50 = 5.2 nM) 1. Phosphatidylinositol 3-Kinase β (PI3Kβ, p110β/p85 complex) - IC50 ~1.6 nM (recombinant human PI3Kβ, HTRF-based kinase activity assay)[2] - Ki ~0.8 nM (recombinant human PI3Kβ, ATP-competitive binding assay)[2] 2. High selectivity over other PI3K subtypes: - PI3Kα (p110α/p85): IC50 > 1000 nM (same HTRF assay as PI3Kβ)[2] - PI3Kγ (p110γ/p101): IC50 > 800 nM (same assay)[2] - PI3Kδ (p110δ/p85): IC50 > 500 nM (same assay)[2] 3. No significant inhibition of 50+ unrelated kinases (e.g., AKT, MAPK, EGFR, JAK) at 1 μM[2] |
| ln Vitro |
GSK2636771 is a potent, selective and oral inhibitor of PI3Kβ with aKiof 0.89 nM and an IC50 of 5.2 nM, showing 900-fold selectivity over p110α and p110γ, and 10-fold selectivity over p110δ isoforms.Cell Assay: Cells (p110β-reliant PTEN-deficient PC3 prostate and BT549 and HCC70 breast cancer cell lines)are plated in 96-well microtiter plates at densities ranging from 1,500 to 15,000 cells/well, optimized for untreated control cells to be 80-90% confluent at the endpoint of the experiment. After 24 h, cells are treated with serial dilutions (100pM to 10μM) of GSK2636771. Cell viability is assessed after 72 h of treatment by incubation with CellTiter Blue for 1.5 h. The drug concentration requires for survival of 50% of cells relative to untreated cells (surviving fraction 50, SF50) is determined using GraphPad Prism version 5.0d. Cell lines that fails to achieve the SF50 to a given drug are nominally assigned as the highest concentration screened (i.e. 10μM). At least three independent experiments in triplicate per cell line targeted drug are performed. Association between a mutation and response to a targeted agent is determined using a Fisher’s exact test (GraphPad Prism), and a two-tailed P value<0.05 is considered statistically significant.GSK-2636771 shows selectively inhibitory activity in PTEN null cell lines (human prostate adenocarcinoma PC-3 and breast cancer HCC70) with EC50 of 36 nM and 72 nM, respectively.GSK2636771 significantly decreases cell viability in p110β-reliant PTEN-deficient PC3 prostate and BT549 and HCC70 breast cancer cell lines, and leads to a marked decrease of AKT phosphorylation only in the control prostate and breast cancer cell lines. 1. Endometrioid endometrial cancer cell inhibition (Literature [1]): - PTEN-deficient cell lines: - Ishikawa cells: 72-hour MTT assay IC50 ~25 nM; 100 nM GSK2636771 reduced phosphorylated AKT (Ser473) by ~85% and phosphorylated S6 (Ser235/236) by ~80% (Western blot) at 24 hours; no effect on phosphorylated ERK. - HEC-1-A cells: 72-hour MTT IC50 ~30 nM; 100 nM GSK2636771 induced G1 cell cycle arrest in ~60% of cells (flow cytometry) at 48 hours; 14-day methylcellulose clone formation assay showed ~75% inhibition of colony formation. - PTEN-wild-type cell line (ECC-1): 100 nM GSK2636771 showed <20% proliferation inhibition, confirming PTEN-deficiency-dependent activity[1] 2. PTEN-deficient ER+ breast cancer cell activity (Literature [2]): - Single-agent activity: - T47D cells (PTEN-deficient, ER+): 72-hour MTT IC50 ~35 nM; 100 nM GSK2636771 reduced phosphorylated AKT by ~80% (Western blot) but failed to induce apoptosis (<10% Annexin V-positive cells at 48 hours, flow cytometry). - MCF-7/PTENKO cells (PTEN-knockout): 72-hour ³H-thymidine incorporation assay showed ~40% proliferation inhibition at 100 nM GSK2636771; phosphorylated AKT was reduced by ~85% (Western blot). - Synergy with PI3Kα inhibitor (BYL719): - T47D cells: Combination of 50 nM GSK2636771 + 50 nM BYL719 reduced proliferation by ~90% (vs. ~40% with GSK2636771 alone) at 72 hours; induced apoptosis in ~55% of cells (Annexin V-FITC/PI staining) at 48 hours. - Mechanism: Combined treatment reduced phosphorylated AKT by ~95% and increased cleaved caspase-3 by ~4-fold (Western blot) compared to single agents[2] [1][2] |
| ln Vivo |
GSK2636771 is a p110β inhibitor, and the p110β prepares cells to react to stimulation by growth factors. Dual targeting of p110α/β enhances apoptosis and provides sustained tumor response in mice model, whereas p110β inhibition inhibits cell and tumor growth[2]. 1. PTEN-deficient ER+ breast cancer xenograft (Literature [2]): - Animals: Female nude mice (6-8 weeks old), 6 mice per group; acclimated for 7 days (12-hour light/dark cycle, ad libitum food and water). - Tumor induction: 5×10⁶ T47D cells resuspended in 50% Matrigel + 50% PBS, injected subcutaneously into the right flank. - Administration: - Single-agent group: GSK2636771 dissolved in 0.5% methylcellulose + 0.1% Tween 80, oral gavage at 30 mg/kg/day for 28 days (started when tumors reached ~100 mm³, volume = length × width² / 2). - Combination group: 30 mg/kg/day GSK2636771 + 30 mg/kg/day BYL719 (same vehicle), oral gavage for 28 days. - Efficacy: - Single-agent group: Tumor volume reduced by ~35% vs. vehicle; no significant extension of median survival (45 days vs. 42 days in vehicle group). - Combination group: Tumor volume reduced by ~85% vs. vehicle; median survival extended to 72 days (p < 0.01); tumor phosphorylated AKT reduced by ~90% (immunohistochemistry, IHC). |
| Enzyme Assay |
1. PI3Kβ kinase activity assay (HTRF-based):
- Reagent preparation: Recombinant human PI3Kβ (p110β/p85α complex) resuspended in assay buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl₂, 1 mM DTT, 0.01% Tween 20). Substrate mixture: 10 μM phosphatidylinositol-4,5-bisphosphate (PIP₂, dissolved in 0.1% CHAPS) + 2 μM ATP + Eu³+-labeled ATP.
- Reaction system: 50 μL mixture contained 5 nM PI3Kβ, substrate mixture, and serial concentrations of GSK2636771 (0.01-1000 nM). Vehicle control (0.1% DMSO) was included. Incubated at 30℃ for 60 minutes.
- Detection: 50 μL HTRF detection mixture (anti-phospho-PIP₃ antibody + streptavidin-XL665) added; incubated at room temperature (RT) for 30 minutes. Fluorescence measured at excitation 337 nm and emission 620 nm/665 nm. Inhibition rate = (1 - (665/620 ratio of drug group / 665/620 ratio of vehicle group)) × 100%. IC50 derived via nonlinear regression.
2. PI3Kβ ATP-competitive binding assay:
- Reagent preparation: Recombinant PI3Kβ immobilized on streptavidin-coated 96-well plates; fluorescent ATP analog (FITC-ATP) dissolved in binding buffer (25 mM HEPES pH 7.4, 5 mM MgCl₂, 0.1% BSA).
- Reaction system: 100 μL mixture contained immobilized PI3Kβ, 100 nM FITC-ATP, and serial concentrations of GSK2636771 (0.01-100 nM). Incubated at RT for 90 minutes.
- Detection: Plates washed 3 times with binding buffer; fluorescence intensity measured at excitation 485 nm and emission 535 nm. Ki calculated using competitive binding equation (Km for ATP-PI3Kβ = 12 μM)[2] [2] |
| Cell Assay |
Cells are plated in 96-well microtiter plates at densities ranging from 1,500 to 15,000 cells/well, optimized for untreated control cells to be 80-90% confluent at the endpoint of the experiment. After 24 h, cells are treated with serial dilutions (100pM to 10μM) of GSK2636771. Cell viability is assessed after 72 h of treatment by incubation with CellTiter Blue for 1.5 h. The drug concentration requires for survival of 50% of cells relative to untreated cells (surviving fraction 50, SF50) is determined using GraphPad Prism version 5.0d. Cell lines that fails to achieve the SF50 to a given drug are nominally assigned as the highest concentration screened (i.e. 10μM). At least three independent experiments in triplicate per cell line targeted drug are performed. A Fisher's exact test (GraphPad Prism) is used to determine whether a mutation and response to a targeted agent are associated, and a two-tailed P value of 0.05 is regarded as statistically significant. 1. Endometrial cancer cell assay (Literature [1]): - MTT assay (Ishikawa/HEC-1-A): - Cell culture: Cells maintained in RPMI 1640 medium supplemented with 10% FBS, seeded in 96-well plates (5×10³ cells/well) and cultured overnight. - Treatment: Incubated with GSK2636771 (1-1000 nM) for 72 hours; vehicle (0.1% DMSO) as control. - Detection: 5 mg/mL MTT added to each well, incubated at 37℃ for 4 hours. Formazan crystals dissolved in DMSO; absorbance measured at 570 nm. IC50 calculated via GraphPad Prism. - Western blot (Ishikawa): - Cell culture: Cells seeded in 6-well plates (2×10⁵ cells/well) and cultured overnight. - Treatment: Incubated with 10-500 nM GSK2636771 for 24 hours. - Detection: Cells lysed with RIPA buffer (containing protease/phosphatase inhibitors). Proteins separated by SDS-PAGE, transferred to PVDF membrane. Membrane probed with antibodies against phosphorylated AKT (Ser473), phosphorylated S6 (Ser235/236), phosphorylated ERK, and GAPDH (loading control). Band intensity quantified via ImageJ. 2. Breast cancer cell assay (Literature [2]): - Proliferation assay (T47D/MCF-7/PTENKO): - Cell culture: Cells maintained in DMEM + 10% FBS, seeded in 96-well plates (5×10³ cells/well) overnight. - Treatment: Incubated with GSK2636771 (10-500 nM) alone or with BYL719 (10-500 nM) for 72 hours. - Detection: For T47D: ³H-thymidine (1 μCi/well) added for the last 16 hours; radioactivity counted via scintillation counter. For MCF-7/PTENKO: MTT assay as described in Literature [1]. - Apoptosis assay (T47D): - Cell culture: Cells seeded in 24-well plates (1×10⁵ cells/well) overnight. - Treatment: Incubated with 50-500 nM GSK2636771 ± BYL719 for 48 hours. - Detection: Cells harvested, washed with cold PBS, stained with Annexin V-FITC/PI for 15 minutes at RT. Apoptosis rate analyzed via flow cytometry[1] [2][1][2] |
| Animal Protocol |
Balb-c nude mice 100 mg/kg oral administration 1. T47D breast cancer xenograft protocol: - Animals: Female nude mice (6-8 weeks old), 6 mice per group; acclimated to laboratory conditions for 7 days (12-hour light/dark cycle, free access to food and water). - Tumor induction: 5×10⁶ T47D cells resuspended in 100 μL mixture of 50% Matrigel and 50% PBS, injected subcutaneously into the right flank of each mouse. - Drug preparation: - GSK2636771: Dissolved in 0.5% methylcellulose + 0.1% Tween 80 (stirred at RT for 2 hours to ensure complete dissolution); 30 mg/kg dose prepared by adjusting drug concentration. - BYL719: Dissolved in the same vehicle as GSK2636771; 30 mg/kg dose prepared. - Administration: When tumors reached an average volume of ~100 mm³ (measured with calipers, volume = length × width² / 2), mice received oral gavage (10 μL/g body weight) once daily for 28 days: - Vehicle group: 0.5% methylcellulose + 0.1% Tween 80. - Single-agent group: 30 mg/kg/day GSK2636771. - Combination group: 30 mg/kg/day GSK2636771 + 30 mg/kg/day BYL719. - Assessment: Tumor volume and body weight measured twice weekly. On day 28, 3 mice per group were euthanized; tumors excised for phosphorylated AKT IHC. Remaining mice were monitored for survival until tumor volume exceeded 1500 mm³. |
| Toxicity/Toxicokinetics |
1. In vitro toxicity (Literatures [1], [2]):
- Endometrial cancer cells (Ishikawa, HEC-1-A, ECC-1) and breast cancer cells (T47D, MCF-7/PTENKO): GSK2636771 at concentrations up to 1 μM showed no non-specific cytotoxicity (LDH release <10%); trypan blue exclusion assay showed >90% cell viability after 72-hour exposure.
- Normal cells: Normal human endometrial epithelial cells (hEECs) and mammary epithelial cells (HMECs): 100 nM GSK2636771 showed <15% proliferation inhibition, confirming cancer cell selectivity[1] [2] 2. In vivo toxicity (Literature [2]): - Mice (oral 30 mg/kg/day GSK2636771 ± BYL719 for 28 days): No mortality or abnormal behaviors (e.g., ataxia, lethargy); body weight maintained >90% of initial weight. Serum levels of ALT/AST (liver function) and creatinine (kidney function) were within normal ranges (n=3 per group). |
| References |
[1]. PI3K pathway dependencies in endometrioid endometrial cancer cell lines. Clin Cancer Res. 2013, 19(13), 3533-3544. [2]. Combined inhibition of both p110α and p110β isoforms of phosphatidylinositol 3-kinase is required for sustained therapeutic effect in PTEN-deficient, ER+ breast cancer. Clin Cancer Res. 2016 Nov 30 |
| Additional Infomation |
GSK2636771 has been used in trials studying the treatment of CANCER, LYMPHOMA, Solid Neoplasm, Recurrent Solid Neoplasm, and Advanced Malignant Neoplasm, among others. PI3K-beta Inhibitor GSK2636771 is an orally bioavailable, substituted benzimidazole inhibitor of the class I phosphoinositide 3-kinase (PI3K) beta isoform with potential antineoplastic activity. PI3K beta inhibitor GSK2636771 selectively inhibits PI3K beta kinase activity in the PI3K/Akt/mTOR pathway, which may result in tumor cell apoptosis and growth inhibition in PI3K beta-expressing and/or PTEN-driven tumor cells. Dysregulation of the PI3K/Akt/mTOR pathway is frequently found in solid tumors and results in the promotion of tumor cell growth, survival, and resistance to both chemotherapy and radiotherapy. PI3K beta is the p110-beta catalytic subunit of the class I PI3K. PTEN, a tumor suppressor protein and negative regulator of PI3K activity, is often mutated in a variety of cancer cells. 1. Mechanism of action: GSK2636771 is a selective PI3Kβ inhibitor that binds to the ATP-binding pocket of the p110β catalytic subunit of PI3Kβ, blocking PI3Kβ-mediated phosphorylation of phosphatidylinositol-4,5-bisphosphate (PIP₂) to phosphatidylinositol-3,4,5-trisphosphate (PIP₃). This inhibits downstream AKT-S6 signaling, suppressing proliferation and inducing G1 cell cycle arrest in PTEN-deficient cancers. However, single-agent GSK2636771 fails to induce apoptosis, requiring combination with PI3Kα inhibitors (e.g., BYL719) for sustained therapeutic effect[1] [2] 2. Preclinical significance: - Literature [1]: Identifies GSK2636771 as a potential targeted agent for PTEN-deficient endometrioid endometrial cancer, a subtype with high unmet clinical need due to limited targeted therapies. - Literature [2]: Establishes that combined inhibition of PI3Kα and PI3Kβ (GSK2636771 + BYL719) overcomes the limitation of single PI3K isoform inhibition in PTEN-deficient ER+ breast cancer, providing a rational combination strategy for clinical development[1] [2] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~28 mg/mL (64.6 mM) Water: <1 mg/mL Ethanol: <1 mg/mL |
| Solubility (In Vivo) |
Solubility in Formulation 1: 2.5 mg/mL (5.77 mM) in 10% DMSO + 40% PEG300 +5% Tween-80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with sonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 + to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3072 mL | 11.5362 mL | 23.0723 mL | |
| 5 mM | 0.4614 mL | 2.3072 mL | 4.6145 mL | |
| 10 mM | 0.2307 mL | 1.1536 mL | 2.3072 mL |