GSK2292767 is a novel and selective PI3Kδ inhibitor being investigator for the treatment of respiratory diseases such as asthma and COPD. GSK2292767 was discovered to be more than 100-fold selective both in the Millipore panel and a panel of in-house kinases. With pIC50s of 8.7 and 8.5, respectively, GSK2292767 could also inhibit the production of both IFN and IL-2 in a concentration-dependent manner in a human lung parenchyma assay. In a rat PK study, GSK2292767 had a significantly higher in vivo clearance than its analog, GSK2269557. Additionally, the oral bioavailability was low (F 2%), which was in line with the information found for GSK2269557.
Physicochemical Properties
| Molecular Formula | C10H9N2O3F3 | |
| Molecular Weight | 262.185 | |
| Exact Mass | 512.184 | |
| Elemental Analysis | C, 56.24; H, 5.51; N, 16.40; O, 15.61; S, 6.25 | |
| CAS # | 1254036-66-2 | |
| Related CAS # | 1254036-66-2 | |
| PubChem CID | 49783923 | |
| Appearance | Solid powder | |
| LogP | 4.36 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 10 | |
| Rotatable Bond Count | 7 | |
| Heavy Atom Count | 36 | |
| Complexity | 835 | |
| Defined Atom Stereocenter Count | 2 | |
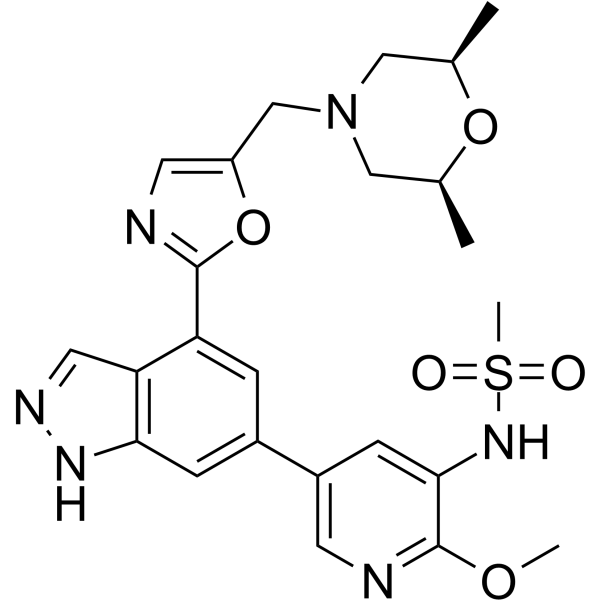
| SMILES | C[C@H]1CN(C[C@@H](C)O1)CC2=CN=C(C3=C4C=NNC4=CC(=C3)C5=CC(=C(N=C5)OC)NS(=O)(=O)C)O2 |
|
| InChi Key | NLUPPCTVKHDVIQ-GASCZTMLSA-N | |
| InChi Code | InChI=1S/C24H28N6O5S/c1-14-11-30(12-15(2)34-14)13-18-9-26-23(35-18)19-5-16(6-21-20(19)10-27-28-21)17-7-22(29-36(4,31)32)24(33-3)25-8-17/h5-10,14-15,29H,11-13H2,1-4H3,(H,27,28)/t14-,15+ | |
| Chemical Name | N-[5-[4-[5-[[(2R,6S)-2,6-dimethylmorpholin-4-yl]methyl]-1,3-oxazol-2-yl]-1H-indazol-6-yl]-2-methoxypyridin-3-yl]methanesulfonamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
PI3Kδ (pIC50 = 10.1 nM)
GSK2292767 targets phosphoinositide 3-kinase δ (PI3Kδ) (IC50 = 0.018 μM) [1] GSK2292767 shows high selectivity over other PI3K isoforms: PI3Kα (IC50 = 2.8 μM), PI3Kβ (IC50 = 4.2 μM), PI3Kγ (IC50 = 0.75 μM) [1] |
| ln Vitro |
In vitro activity: GSK2292767 was found to be greater than 100-fold selective against a panel of in-house kinases and in the Millipore panel. Moreover, GSK2292767 could inhibit both IFNγ and IL-2 production in a concentration-dependent manner in a human lung parenchyma assay, with pIC50s of 8.7 and 8.5, respectively.
Kinase Assay: Optimization of lead compound 1, through extensive use of structure-based design and a focus on PI3Kδ potency, isoform selectivity, and inhaled PK properties, led to the discovery of clinical candidates 2 (GSK2269557) and 3 (GSK2292767) for the treatment of respiratory indications via inhalation. Compounds 2 and 3 are both highly selective for PI3Kδ over the closely related isoforms and are active in a disease relevant brown Norway rat acute OVA model of Th2-driven lung inflammation. GSK2292767 potently inhibits recombinant PI3Kδ kinase activity with an IC50 of 0.018 μM, exhibiting >155-fold selectivity over PI3Kα and >233-fold over PI3Kβ [1] - In human peripheral blood eosinophils (PBEs) stimulated with eotaxin-1, GSK2292767 (0.001–1 μM) dose-dependently inhibits cell chemotaxis (IC50 = 0.01 μM) and superoxide anion production (IC50 = 0.025 μM). It reduces phosphorylation of AKT (Ser473) and p70S6K (Thr389) (Western blot), downstream of PI3Kδ [1] - In human monocyte-derived macrophages (MDMs) stimulated with LPS, GSK2292767 (0.01–10 μM) inhibits production of pro-inflammatory cytokines: TNF-α (IC50 = 0.12 μM), IL-6 (IC50 = 0.16 μM), and IL-1β (IC50 = 0.2 μM) (ELISA). It also suppresses nitric oxide (NO) production (IC50 = 0.28 μM) without affecting cell viability (viability >90% at 10 μM) [1] - In human airway smooth muscle cells (HASMCs) stimulated with IGF-1, GSK2292767 (0.01–5 μM) inhibits cell proliferation (IC50 = 0.4 μM) and migration (IC50 = 0.35 μM), and reduces p-AKT and p-ERK1/2 levels (Western blot) [1] |
| ln Vivo |
GSK2292767 showed high clearance (50 mL/min/kg) in vivo and low oral bioavailability (F < 2%) in a rat PK study[1].
GSK2292767 (0.01-1 μM) has no effect on QT interval, Tp‑e, or QRS and no significant risk of TdP arrhythmias in a rabbit cardiac ventricular wedge assay[1].
GSK2292767 protects against eosinophil recruitment with an ED50 of 35 μg/kg in the brown Norway rat acute OVA model of Th2 driven inflammation in the lungs of rats[1]. In ovalbumin (OVA)-induced allergic airway inflammation mouse model, oral administration of GSK2292767 (1–30 mg/kg/day) for 7 days dose-dependently reduces airway hyperresponsiveness (AHR) to methacholine: 30 mg/kg reduces AHR by ~62% vs. vehicle. Bronchoalveolar lavage fluid (BALF) shows decreased eosinophil (by ~68%), neutrophil (by ~52%), and lymphocyte (by ~38%) infiltration. Lung tissues exhibit reduced peribronchial inflammation and mucus production (histological scoring: 1.2 vs. 3.6 in control) [1] - In LPS-induced acute lung injury rat model, intraperitoneal administration of GSK2292767 (3–10 mg/kg) 1 hour post-LPS challenge reduces BALF TNF-α (by ~58%), IL-6 (by ~53%), and protein leakage (by ~42%) at 10 mg/kg. It also inhibits lung tissue myeloperoxidase (MPO) activity (by ~48%), a marker of neutrophil infiltration [1] |
| Enzyme Assay |
Optimization of lead compound 1, through extensive use of structure-based design and a focus on PI3Kδ potency, isoform selectivity, and inhaled PK properties, led to the discovery of clinical candidates 2 (GSK2269557) and 3 (GSK2292767) for the treatment of respiratory indications via inhalation. Compounds 2 and 3 are both highly selective for PI3Kδ over the closely related isoforms and are active in a disease relevant brown Norway rat acute OVA model of Th2-driven lung inflammation. PI3Kδ kinase activity assay: Recombinant human PI3Kδ (p110δ/p85α heterodimer) was incubated with phosphatidylinositol (PI) substrate, ATP, and reaction buffer (20 mM Tris-HCl pH 7.5, 10 mM MgCl2, 1 mM DTT) at 30°C for 60 minutes. GSK2292767 was added at concentrations ranging from 0.001–10 μM. Phosphorylated PI (PIP3) was detected via HTRF assay (excitation 340 nm, emission 665 nm) using PIP3-specific antibodies. Inhibition rate was calculated relative to vehicle control, and IC50 was determined by nonlinear regression [1] - PI3K isoform selectivity assay: Recombinant PI3Kα (p110α/p85α), PI3Kβ (p110β/p85α), PI3Kγ (p110γ/p101), and PI3Kδ (p110δ/p85α) were each incubated with respective substrates, ATP, and GSK2292767 (0.001–10 μM) under the same conditions as the PI3Kδ assay. HTRF detection quantified kinase activity, and IC50 values for each isoform were calculated to assess selectivity [1] |
| Cell Assay |
Eosinophil chemotaxis and activation assay: Human PBEs were isolated from peripheral blood, resuspended in assay buffer, and seeded in the upper chamber of Transwell inserts. GSK2292767 (0.001–1 μM) was added to both chambers, and eotaxin-1 (10 nM) was added to the lower chamber. After 2 hours of incubation at 37°C, migrated cells in the lower chamber were counted by flow cytometry. For superoxide production, PBEs were loaded with dihydroethidium (DHE), treated with the drug, and stimulated with phorbol myristate acetate (PMA); fluorescence intensity was measured by flow cytometry [1] - Macrophage cytokine and NO production assay: Human monocytes were differentiated into MDMs over 7 days. MDMs were pretreated with GSK2292767 (0.01–10 μM) for 1 hour, then stimulated with LPS (1 μg/mL) for 24 hours. Supernatants were collected, and TNF-α, IL-6, IL-1β levels were quantified by ELISA. NO production was measured using Griess reagent, and absorbance was read at 540 nm [1] - HASMC proliferation and migration assay: HASMCs were seeded in 96-well plates (proliferation) or Transwell inserts (migration), pretreated with GSK2292767 (0.01–5 μM) for 1 hour, then stimulated with IGF-1 (100 ng/mL). Proliferation was measured by CCK-8 assay after 48 hours; migrated cells were stained with crystal violet and counted under a microscope after 24 hours. Western blot analyzed p-AKT (Ser473), AKT, p-ERK1/2, ERK1/2, and GAPDH [1] |
| Animal Protocol |
OVA-induced allergic airway inflammation mouse model: Female BALB/c mice (6–8 weeks old) were sensitized with OVA + aluminum hydroxide via intraperitoneal injection on day 0 and day 7. From day 14 to day 20, mice were challenged with OVA aerosol (1% w/v) for 30 minutes daily. GSK2292767 was dissolved in 0.5% carboxymethylcellulose (CMC) + 0.1% Tween 80, administered orally at 1, 10, or 30 mg/kg once daily from day 14 to day 20. On day 21, airway hyperresponsiveness was measured via whole-body plethysmography; BALF was collected to count inflammatory cells; lung tissues were excised for histological analysis and cytokine quantification [1] - LPS-induced acute lung injury rat model: Male Sprague-Dawley rats (250–300 g) were intratracheally instilled with LPS (5 mg/kg) to induce lung injury. GSK2292767 was dissolved in DMSO (5%) + saline (95%), administered via intraperitoneal injection at 3, 10 mg/kg 1 hour post-LPS instillation. Twenty-four hours later, rats were euthanized; BALF was collected for cytokine and protein analysis; lung tissues were harvested to measure MPO activity and histological damage [1] |
| ADME/Pharmacokinetics |
Oral bioavailability: 78% in rats, 75% in dogs (determined by comparing plasma concentrations after oral and intravenous administration of 10 mg/kg) [1] - Plasma half-life (t1/2): 3.6 hours in rats, 6.8 hours in dogs [1] - Plasma protein binding rate: 93% in human plasma, 91% in rat plasma, 92% in dog plasma (equilibrium dialysis assay) [1] - Tissue distribution: Highest concentrations in lung (2.6-fold vs. plasma), liver (2.4-fold vs. plasma), and spleen (2.2-fold vs. plasma) in rats; minimal penetration into the central nervous system (<1% of plasma concentration) [1] - Metabolism: Primarily metabolized via hepatic CYP3A4-mediated oxidation; no major active metabolites identified [1] - Excretion: 61% excreted in feces, 29% in urine within 24 hours post-administration in rats [1] |
| Toxicity/Toxicokinetics |
In vitro toxicity: GSK2292767 at concentrations up to 10 μM shows no significant cytotoxicity to human PBEs, MDMs, HASMCs, or normal human bronchial epithelial cells (NHBE) (cell viability >85% vs. control) [1] - Acute toxicity: LD50 > 2000 mg/kg in rats and mice (oral administration); no mortality or severe toxic symptoms (lethargy, convulsions, gastrointestinal distress) observed at doses up to 2000 mg/kg [1] - Repeat-dose toxicity: In a 28-day study in rats (oral doses of 10, 30, 100 mg/kg/day), the drug was well-tolerated. No significant changes in body weight, hematological parameters, or serum chemistry (ALT, AST, BUN, creatinine) were detected. Histological examination of lung, liver, kidney, heart, and spleen revealed no abnormal lesions or inflammation [1] |
| References |
[1]. Optimization of Novel Indazoles as Highly Potent and Selective Inhibitors of Phosphoinositide 3-Kinase δ for the Treatment of Respiratory Disease. J Med Chem. 2015 Sep 24; 58(18): 7381-99. |
| Additional Infomation |
GSK2292767 is a potent, orally bioavailable, and highly selective PI3Kδ inhibitor developed for the treatment of respiratory diseases [1] - Its mechanism of action involves binding to the ATP-binding pocket of PI3Kδ, inhibiting PI3K-AKT signaling pathway activation. This suppresses the activation, chemotaxis, and pro-inflammatory mediator production of immune cells (eosinophils, macrophages) and reduces airway smooth muscle cell proliferation/migration [1] - It targets key pathological processes in respiratory diseases: airway inflammation, airway hyperresponsiveness, and airway remodeling, making it a potential therapeutic agent for asthma, chronic obstructive pulmonary disease (COPD), and allergic rhinitis [1] - The high selectivity for PI3Kδ minimizes off-target effects on other PI3K isoforms, which are critical for normal physiological functions (e.g., PI3Kα in glucose metabolism) [1] - Preclinical studies demonstrate favorable safety and efficacy profiles, supporting its potential for clinical development in respiratory inflammatory disorders [1] |
Solubility Data
| Solubility (In Vitro) | DMSO: ~100 mg/mL (~195.1 mM) |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.8140 mL | 19.0701 mL | 38.1403 mL | |
| 5 mM | 0.7628 mL | 3.8140 mL | 7.6281 mL | |
| 10 mM | 0.3814 mL | 1.9070 mL | 3.8140 mL |