The cinnamoylmorphinone, clocinnamox, has recently been characterized in tests of antinociception in mice and monkeys (Comer et al. 1992; Burke et al. 1994; Zernig et al. 1994), and was described as an irreversible opioid antagonist with relative selectivity for g- over ~¢- and 0-opioid receptors. An advantage of clocinnamox relative to [3-FNA in behavioral studies is that the former does not have an initial, probably ~:-receptor mediated (e.g., Ward et al. 1982) agonist effect which limits behavioral testing soon (i.e., <24 h) after [3-FNA administration. Burke et al. (1994) reported a correspondence between the antagonist effects of clocinnamox against the antinociceptive effects of morphine in mice and an observed decrease in the number of g-receptor binding sites in mouse brain. Thus, clocinnamox (IR 3.2 mg/kg) produced an almost total suppression in [3H]DAMGO ([D-Ala 2, N-Me-Phe 4, glycolSl-enkephalin) binding sites in mouse brain, peaking 1 h after administration, and the number of sites substantially recovered by 8 days after administration. At this dose, clocinnamox caused a large right and downward shift in the morphine dose-effect curve in thermal antinociception, and clocinnamox did not have an antinociceptive effect by itself. The time course of the suppression of morphine's antinociceptive effects mirrored well the decrease in [3H]DAMGO binding sites (Burke et al. 1994). More recent in vitro and ex vivo experiments in mice confirmed that clocinnamox exhibited wash-resistant binding at g-, but not ~;- or 0-, receptors, thus supporting the in vivo selectivity of its antagonist effects (Zernig et al. 1994, 1995; Broadbear et al., manuscript in preparation). In the present studies, we examined whether clocinnamox presented an opioid antagonist profile in a non-antinociceptive behavioral assay in rhesus monkeys, i.e., suppression of schedule-controlled fo0d-reinforced responding. This procedure is a simple means to study the pharmacology of bt- (e.g., etonitazene and fentanyl), ~:- (e.g., U69,593), and 0- (e.g., BW373U86) opioid agonists in vivo in rhesus monkeys (Negus et al. 1993, 1994), and is especially helpful in evaluating clocinnamox's bt- over 0-selectivity, given that the only nonpeptide O-agonist characterized to date, BW373U86, is inactive as a thermal analgesic in rhesus monkeys, but does suppress food-reinforced responding through a 0- receptor mechanism (see Negus et al. 1994; Butelman et al. 1995b) [2].
Physicochemical Properties
| Molecular Formula | C30H33CLN2O7S |
| Molecular Weight | 601.11 |
| Exact Mass | 600.17 |
| Elemental Analysis | C, 59.94; H, 5.53; Cl, 5.90; N, 4.66; O, 18.63; S, 5.33 |
| CAS # | 117332-69-1 |
| Related CAS # | 122566-12-5 |
| PubChem CID | 6438373 |
| Appearance | Typically exists as solid at room temperature |
| Boiling Point | 753.4ºC at 760mmHg |
| Flash Point | 409.4ºC |
| Vapour Pressure | 1.74E-23mmHg at 25°C |
| LogP | 4.929 |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 8 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 41 |
| Complexity | 1040 |
| Defined Atom Stereocenter Count | 4 |
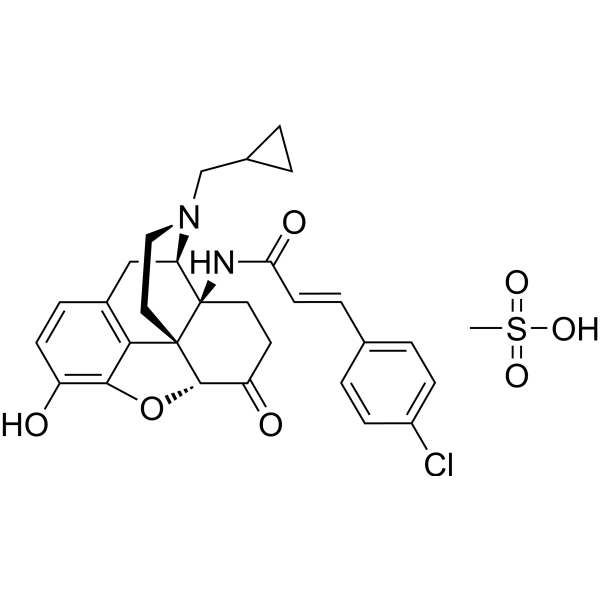
| SMILES | CS(=O)(O)=O.ClC1C=CC(/C=C/C(N[C@@]23CCC(=O)[C@H]4[C@]52CCN(CC2CC2)[C@@H]3CC2=C5C(O4)=C(C=C2)O)=O)=CC=1 |
| InChi Key | XAXNKAGAUFXFDO-JVJDXIHNSA-N |
| InChi Code | InChI=1S/C29H29ClN2O4.CH4O3S/c30-20-7-3-17(4-8-20)5-10-24(35)31-29-12-11-22(34)27-28(29)13-14-32(16-18-1-2-18)23(29)15-19-6-9-21(33)26(36-27)25(19)28;1-5(2,3)4/h3-10,18,23,27,33H,1-2,11-16H2,(H,31,35);1H3,(H,2,3,4)/b10-5+;/t23-,27+,28+,29-;/m1./s1 |
| Chemical Name | (E)-N-[(4R,4aS,7aR,12bR)-3-(cyclopropylmethyl)-9-hydroxy-7-oxo-2,4,5,6,7a,13-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-4a-yl]-3-(4-chlorophenyl)prop-2-enamide;methanesulfonic acid |
| Synonyms | Methoclocinnamox; 117332-69-1; Clocinnamox mesylate; Clocinnamox (mesylate); C-CAM; (E)-N-[(4R,4aS,7aR,12bR)-3-(cyclopropylmethyl)-9-hydroxy-7-oxo-2,4,5,6,7a,13-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-4a-yl]-3-(4-chlorophenyl)prop-2-enamide;methanesulfonic acid; 14b-(p-Chlorocinnamoylamino)-7,8-dihydro-N-cyclopropylmethylmorphinone mesylate; 14-beta-(4-Chlorocinnamoylamino)-7,8-dihydro-N-cyclopropylmethylnorpmorphine mesylate; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | Opioid mu, kappa and delta-receptors |
| ln Vivo |
The warm water (55 degrees C) tail-withdrawal procedure was used to assess the analgesic effects of the prototypic mu opioid agonists, morphine and fentanyl, in mice. Both drugs produced full analgesic effects under these conditions, which were dose-dependently antagonized by naltrexone. The pA2 values for naltrexone with morphine and with fentanyl were not significantly different. Low doses (e.g., 0.32 mg/kg) of Clocinnamox [C-CAM.14 beta-(p- chlorocinnamoylamino)-7,8-dihydro-N-cyclopropylmethylnormorphin one mesylate] produced rightward shifts in the dose-effect curves for each drug, whereas high doses (e.g., 32 mg/kg) depressed the maximal analgesic response. In addition, it was observed that higher doses of C-CAM were required to produce a shift down in the fentanyl dose-effect curve than were required to produce a shift down in the morphine dose-effect curve, which suggests that fentanyl is more efficacious than morphine. The highest dose of C-CAM (32 mg/kg) antagonized the analgesic effect of morphine for up to 8 days. In contrast, the antagonist activity of naltrexone (100 mg/kg) against morphine lasted for only 2 days. Finally, when naloxone was administered simultaneously with 32 mg/kg C-CAM 2 days before determination of the morphine dose-effect function, the antagonist effect of C-CAM was prevented in a dose-dependent manner. Taken together, these results suggest that C-CAM may be producing its antagonist action at opioid receptors through a nonequilibrium mechanism [1].
The antagonist effects of Clocinnamox were evaluated against opioid agonists, acting at mu, kappa and delta-receptors, in rhesus monkeys (n = 3-4) responding under a fixed-ratio 30 (FR 30) schedule for food delivery. Clocinnamox (0.032-0.1 mg/kg) dose-dependently antagonized fentanyl (0.001-0.32 mg/kg) after either a 3-h or 1-day pretreatment; there was substantial recovery of agonist potency by 1 week after Clocinnamox. Etonitazene (0.0001-0.01 mg/kg) was also antagonized by Clocinnamox (0.1 mg/kg), but to a lesser extent than fentanyl. The smaller extent of antagonism was not due to the appearance of non mu-opioid response-decreasing effects of etonitazene, since the competitive antagonist quadazocine (0.1 mg/kg) shifted the etonitazene dose-effect curve in the presence of Clocinnamox (0.1 mg/kg). Clocinnamox (0.1-0.32 mg/kg) did not antagonize the rate-suppressing effects of the delta-agonist BW373U86 (0.0.01-1.0 mg/kg) or the kappa-agonist U69,593 (0.001-0.032 mg/kg). These results are consistent with previous in vivo and in vitro evidence that characterized clocinnamox as an insurmountable antagonist, with selectivity for mu- over kappa- and delta-receptors [2]. |
| Animal Protocol |
Test sessions [2] During test sessions, agonist doses were administered at the beginning of each cycle, using a cumulative dosing procedure, increasing by 1/2 log unit steps. Dosing continued until responding was eliminated in each subject, or after the appearance of untoward effects, such as preconvulsant behavior in the presence of BW373U86. Dose-effect curves were determined for etonitazene, fentanyl, BW373U86 and U69,593. These dose-effect curves were redetermined 3 h (fentanyl only), 1 day and 1-2 weeks after pre321 treatment with single Clocinnamox doses (0.032-0.32 mg/kg, n=3-4). In another experiment, the etonitazene dose-effect curve was also redetermined 30 rain after pretreatment with quadazocine (0.1 mg/kg), either alone or 1 day after Clocinnamox pretreatment (0.1 mg/kg). Clocinnamox mesylate was dissolved in sterile water and injected SC in the thigh or the back, typically at volumes of 0.1 ml/kg body weight. All drug doses were expressed as salt weights. |
| References |
[1]. Clocinnamox: a novel, systemically-active, irreversible opioid antagonist. J Pharmacol Exp Ther. 1992, 262, 3. [2]. Clocinnamox antagonism of opioid suppression of schedule-controlled responding in rhesus monkeys. Psychopharmacology (Berl). 1996, 123, 4. |
| Additional Infomation | An interesting aspect of the g-antagonist effects of Clocinnamox (0.1 mg/kg, 24 h after administration) in the present studies is that it apparently produced a larger shift of fentanyl's dose-effect curve compared to etonitazene's dose-effect curve. It has previously been suggested that varying agonist efficacy results in differential sensitivity to irreversible antagonism. For example, the g-selective irreversible antagonist, ~3-FNA, produced larger shifts in the antinociceptive dose-effect curves of agonists thought to have low efficacy, relative to agonists thought to have high efficacy. This trend was observed in rodents in a variety of assays (tail flick, hot plate, abdominal constriction) following either central or systemic administration (Zimmerman et al. 1987; Adams et al. 1990; Mjanger and Yaksh 1991). The present data therefore suggest that fentanyl has lower efficacy than etonitazene in this procedure. Etonitazene has previously been described as having higher efficacy than the fentanyl congener, alfentanil, in thermal antinociception in rhesus monkeys (Walker et al. 1995). Relevant to this issue, quadazocine (0.1 mg/kg) pretreatment caused a further shift in the etonitazene dose-effect curve in the presence of Clocinnamox, of similar magnitude than observed in the absence of clocinnamox. This quadazocine pretreatment dose (0.1 mg/kg) would not antagonize the effects of 0- or ~:-agonists in this procedure (see Negus et al. 1994). Therefore, the extent of the clocinnamox-induced shift in etonitazene's dose-effect curve was not limited by the appearance of non B-opioid receptor-mediated effects. An alternative explanation for clocinnamox's differential shift of the etonitazene and fentanyl dose-effect curves is the possibility that these compounds had differential affinity for putative g-receptor subtypes (e.g., Pasternak 1993). This explanation cannot be evaluated at present, due to a lack of information on B-subtype pharmacology in primates. The present data show that Clocinnamox acts as a relatively long-lasting, B-selective antagonist in rhesus monkeys in a non-antinociceptive endpoint, and that it is devoid of an initial agonist effect. These findings extend previous data obtained in the procedure with the competitive antagonists quadazocine and naltrindole (Negus et al. 1993, 1994). Thus Clocinnamox, which shows insurmountable antagonist effects in tests of antinociception in primates (Zernig et al. 1993), can be studied in this simple operant procedure, which is sensitive to a variety of opioids that may be inactive in other behavioral endpoints, including tests of antinociception. |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.6636 mL | 8.3179 mL | 16.6359 mL | |
| 5 mM | 0.3327 mL | 1.6636 mL | 3.3272 mL | |
| 10 mM | 0.1664 mL | 0.8318 mL | 1.6636 mL |