Physicochemical Properties
| Molecular Formula | C13H13NO4 |
| Molecular Weight | 247.24662 |
| Exact Mass | 247.084 |
| CAS # | 179067-99-3 |
| PubChem CID | 6278000 |
| Appearance | White to off-white solid powder |
| LogP | 1.9 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 3 |
| Heavy Atom Count | 18 |
| Complexity | 395 |
| Defined Atom Stereocenter Count | 0 |
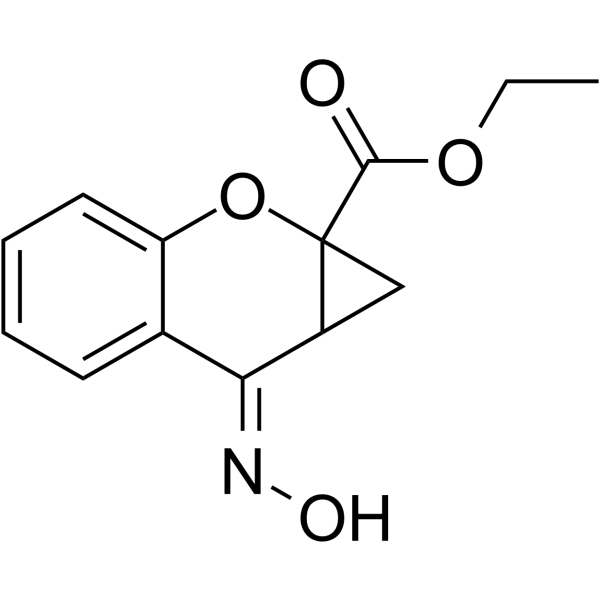
| SMILES | CCOC(C12CC1/C(/C1=CC=CC=C1O2)=N/O)=O |
| InChi Key | FXCTZFMSAHZQTR-SDNWHVSQSA-N |
| InChi Code | InChI=1S/C13H13NO4/c1-2-17-12(15)13-7-9(13)11(14-16)8-5-3-4-6-10(8)18-13/h3-6,9,16H,2,7H2,1H3/b14-11+ |
| Chemical Name | ethyl (7Z)-7-hydroxyimino-1,7a-dihydrocyclopropa[b]chromene-1a-carboxylate |
| Synonyms | CPCCOEt; 179067-99-3; CHEMBL327783; ethyl (7E)-7-hydroxyimino-1,7a-dihydrocyclopropa[b]chromene-1a-carboxylate; Benzo[b]cyclopropa[e]pyran-1a(1H)-carboxylic acid, 7,7a-dihydro-7-(hydroxyimino)-, ethyl ester; C13H13NO4; SCHEMBL12648369; HMS3412A05; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | mGluR1b/metabotropic glutamate receptor 1b |
| ln Vitro |
In transfected CHO cells expressing the human metabotropic glutamate receptor mGlu1alpha, 7-(hydroxyimino)cyclopropan[b]-chromen-1a-carboxylic acid ethylester (CPCCOEt) was found to antagonize L-quisqualate-induced phosphoinositide hydrolysis in a non-competitive and reversible manner (apparent pKi value, 4.76+/-0.18; n=3). This suggests that CPCCOEt antagonizes type 1alpha metabotropic glutamate receptor activation by interacting with a site distinct from the agonist binding site[2].
As shown in Fig. 1 (panel A), increasing concentrations of CPCCOEt dramatically inhibited l-quisqualate-induced [3H]InsP accumulation in IPTG-induced CHO–Lac–mGlu1α cells. CPCCOEt not only resulted in a progressive rightward-shift in the concentration–response curve to l-quisqualate, but it also caused a dramatic decrease in the maximal response stimulated by the agonist, revealing that antagonism was not competitive. A significant effect on the l-quisqualate-stimulated response was observed at 3 μM CPCCOEt and complete inhibition of mGlu1α receptor-mediated signalling was observed in the presence of 300 μM CPCCOEt. Under the same experimental conditions, the prototypic mGlu receptor antagonist (S)-MCPG was found to behave as a competitive antagonist causing a rightward-shift in the concentration–response curve without decreasing the maximal response to l-quisqualate (Fig. 1 panel A, inset, pKB, 3.73±0.05). Concentration–response curves were fitted to provide estimates of the maximal response at each concentration of CPCCOEt and an apparent Ki value assuming non-competitive antagonism was estimated using the following equation: where C is a given concentration of antagonist and Rmax and Rp are the maximal responses obtained in the absence or in the presence of this concentration of the antagonist, respectively (Dixon and Webb, 1979). Using this equation, the estimated pKi value for CPCCOEt was 4.76±0.18 (range 4.33–5.05 for 12 estimates from three independent experiments). This value is in accordance with the values reported previously (Annoura et al., 1996, Casabona et al., 1997), although the estimates made in these studies were obtained assuming competitive inhibition.[1] The activity and selectivity towards mGlu1 receptors suggests that the antagonist CPCCOEt constitutes a particularly interesting pharmacological tool for the study of glutamatergic transmission through mGlu receptors (Annoura et al., 1996, Casabona et al., 1997). However, until now the antagonist action of CPCCOEt has only been assessed by employing a single stimulatory concentration of agonist in the presence of increasing concentrations of antagonist, and thus the nature of the antagonism had not been assessed. The non-competitive profile of CPCCOEt against l-quisqualate or glutamate (data not shown) suggests that it interacts with a site distinct from the agonist binding site to interfere with the functional activation of the receptor. Accordingly, Okamoto et al. (1998)have recently reported an inability of CPCCOEt to displace specific [3H]quisqualate binding from membranes of Sf9 cells infected to express the mGlu1 receptor. One of the characteristics of mGlu receptors is their particularly large N-terminal extracellular domain wherein is located the agonist binding site (Conn and Pin, 1997, Okamoto et al., 1998), contrasting thereby with other G-protein-coupled receptors where agonist binding is more likely to involve membrane proximal domains. Therefore the use of this antagonist in functional studies would probably help in delineating the structural determinants and the molecular mechanisms of the activation of this particular family of receptors. As the inhibitory effect of a non-competitive antagonist cannot be surmounted by increasing concentrations of agonists, such a compound may constitute a valuable tool for pharmacological studies. This is particularly the case for glutamate receptor antagonists as there is substantial variation in the endogenously generated concentration of glutamate in tissue preparations. Thus, the non-competitive antagonism of CPCCOEt may prove extremely useful in in vivo or in vitro studies where a competitive antagonist might vary in its effectiveness. Thus, the selectivity, high activity and the non-competitive, but reversible, properties of CPCCOEt at the mGlu1 receptor make it a highly valuable and complementary tool for the study of the role of this important mGlu receptor in glutamatergic transmission in the CNS. [1] |
| Enzyme Assay | Metabotropic glutamate receptors (mGluRs) are a family of G protein-coupled receptors characterized by a large, extracellular N-terminal domain comprising the glutamate-binding site. In the current study, we examined the pharmacological profile and site of action of the non-amino-acid antagonist 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester (CPCCOEt). CPCCOEt selectively inhibited glutamate-induced increases in intracellular calcium at human mGluR1b (hmGluR1b) with an apparent IC50 of 6.5 microM while having no agonist or antagonist activity at hmGluR2, -4a, -5a, -7b, and -8a up to 100 microM. Schild analysis indicated that CPCCOEt acts in a noncompetitive manner by decreasing the efficacy of glutamate-stimulated phosphoinositide hydrolysis without affecting the EC50 value or Hill coefficient of glutamate. Similarly, CPCCOEt did not displace [3H]glutamate binding to membranes prepared from mGluR1a-expressing cells. To elucidate the site of action, we systematically exchanged segments and single amino acids between hmGluR1b and the related subtype, hmGluR5a. Substitution of Thr815 and Ala818, located at the extracellular surface of transmembrane segment VII, with the homologous amino acids of hmGluR5a eliminated CPCCOEt inhibition of hmGluR1b. In contrast, introduction of Thr815 and Ala818 at the homologous positions of hmGluR5a conferred complete inhibition by CPCCOEt (IC50 = 6.6 microM), i.e., a gain of function. These data suggest that CPCCOEt represents a novel class of G protein-coupled receptor antagonists inhibiting receptor signaling without affecting ligand binding. We propose that the interaction of CPCCOEt with Thr815 and Ala818 of mGluR1 disrupts receptor activation by inhibiting an intramolecular interaction between the agonist-bound extracellular domain and the transmembrane domain[1]. |
| Cell Assay | In order to assess the reversibility of the antagonism, the cells were preincubated for 30 min in the absence or presence of 100 μM CPCCOEt, and then washed three times with KHB. The response to a subsequent challenge of the cells with l-quisqualate (10 μM) was found not to be significantly different from that measured in cells not exposed to the antagonist, indicating that the inhibitory effect of CPCCOEt was fully reversible (Fig. 1 panel B). In order to investigate the specificity of the non-competitive antagonism of CPCCOEt for mGlu receptors, the effect of 100 μM CPCCOEt was measured on (−)-norepinephrine- and methacholine-induced phosphoinositide hydrolysis in transfected CHO cells expressing α1A-adrenoceptors (receptor density, 5 pmol mg−1 protein; EC50 for (−)-norepinephrine, 0.2 μM), or M3-muscarinic receptors (3 pmol mg−1 protein; EC50 for methacholine, 1.8 μM), respectively. As shown in Fig. 1 panel C, CPCCOEt had no effect on the agonist-stimulated response in these two model cell-lines. [2] |
| References |
[1]. CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol Pharmacol. 1999 Mar;55(3):453-61. [2]. Reversible and non-competitive antagonist profile of CPCCOEt at the human type 1alpha metabotropic glutamate receptor. Neuropharmacology. 1998 Dec;37(12):1645-7. |
| Additional Infomation | Glutamate metabotropic (mGlu) receptors are classified in three groups based on structural homologies, signal transduction mechanisms and pharmacological properties (Conn and Pin, 1997). Initially, the search for new potent and selective agonists or antagonists of metabotropic receptors led to the development of a variety of compounds, notably the phenylglycines (Watkins and Collingridge, 1994). However, only very recently have compounds capable of distinguishing between mGlu receptor subtypes within a given group been reported. Thus, a small number of compounds can distinguish between the group I mGlu1 and mGlu5 receptors. These include 1-aminoindan-1,5-dicarboxylic acid (AIDA, Moroni et al., 1997), (RS)-2-chloro-5-hydroxyphenylglycine (CHPG, Doherty et al., 1997), and 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate (CPCCOEt; Annoura et al., 1996), the latter exhibiting the greatest activity and selectivity for antagonism of the mGlu1 (over mGlu5) receptor (Casabona et al., 1997). In the present study, the mechanism by which CPCCOEt inhibits mGlu1 receptor signalling was further investigated by assessing the ability of this compound to inhibit l-quisqualate-stimulated phosphoinositide hydrolysis in CHO cells expressing the recombinant human glutamate mGlu1α receptor (Hermans et al., 1998). [1] |
Solubility Data
| Solubility (In Vitro) | DMSO: 100 mg/mL (404.45 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (10.11 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2.5 mg/mL (10.11 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (10.11 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.0445 mL | 20.2224 mL | 40.4449 mL | |
| 5 mM | 0.8089 mL | 4.0445 mL | 8.0890 mL | |
| 10 mM | 0.4044 mL | 2.0222 mL | 4.0445 mL |