CH7057288 is a novel, potent and selective inhibitor of TRK (tyrosine receptor kinase) with IC50 values of 1.1 nM, 7.8 nM and 5.1 nM for TRKA, TRKB, and TRKC respectively. In cell-free kinase tests, CH7057288 demonstrated specific inhibitory activity against TRKA, TRKB, and TRKC. It also inhibited the growth of TRK fusion-positive cell lines, but not that of TRK-negative cell lines. Significant tumor growth inhibition was seen in xenograft models of TRK fusion-positive cells that were implanted subcutaneously. Moreover, in an intracranial implantation model that replicates brain metastasis, CH7057288 significantly increased event-free survival and caused intracranial tumors to recede. In patients exhibiting disease progression following treatment with a TRK inhibitor in clinical development, resistant mutations in TRK have recently been reported. In vitro and in vivo activity levels of CH7057288 against some of the resistant mutants were comparable to those observed with wild-type TRK. In conclusion, CH7057288 shows promise as a treatment for cancers that fuse the TRK gene. Lung and colorectal cancer are among the many cancer types in which TRK receptor tyrosine kinases are expressed as fusion proteins encoded by different fusion genes. Due to their strong carcinogenic properties, these fusion proteins are considered to be promising targets for therapy.
Physicochemical Properties
| Molecular Formula | C32H31N3O5S |
| Molecular Weight | 569.670646905899 |
| Exact Mass | 569.2 |
| Elemental Analysis | C, 67.47; H, 5.49; N, 7.38; O, 14.04; S, 5.63 |
| CAS # | 2095616-82-1 |
| Related CAS # | 2095616-82-1 |
| PubChem CID | 131839646 |
| Appearance | Off-white to yellow solid powder |
| LogP | 5.3 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 41 |
| Complexity | 1210 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | DCGOHGQJHJXBGW-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C32H31N3O5S/c1-18-14-20(30(37)34-31(2,3)4)16-21(33-18)10-8-19-9-12-24-26(15-19)40-29-27(24)28(36)23-13-11-22(35-41(7,38)39)17-25(23)32(29,5)6/h9,11-17,35H,1-7H3,(H,34,37) |
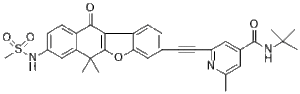
| Chemical Name | N-tert-butyl-2-[2-[8-(methanesulfonamido)-6,6-dimethyl-11-oxonaphtho[2,3-b][1]benzofuran-3-yl]ethynyl]-6-methylpyridine-4-carboxamide |
| Synonyms | CH7057288; CH-7057288; CH 7057288 |
| HS Tariff Code | 2934.99.03.00 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
TrkA (IC50 = 1.1 nM); TrkC (IC50 = 5.1 nM); TrkB (IC50 = 7.8 nM) CH7057288 inhibits the growth of TRK fusion-positive cell lines, but not that of TRK-negative cell lines, and exhibits selective inhibitory activity against TRKA, TRKB, and TRKC in cell-free kinase assays. As a downstream signaling of TRK fusion, CH7057288 inhibits the E2F and mitogen-activated protein kinase (MAPK) pathways[1]. |
| ln Vitro |
CH7057288 inhibits the growth of TRK fusion-positive cell lines, but not that of TRK-negative cell lines, and exhibits selective inhibitory activity against TRKA, TRKB, and TRKC in cell-free kinase assays. As a downstream signaling of TRK fusion, CH7057288 inhibits the E2F and mitogen-activated protein kinase (MAPK) pathways[1]. CH7057288 potently inhibited autophosphorylation of TRK in TRK fusion-positive cancer cell lines (CUTO-3, KM12-Luc, MO-91) in a dose-dependent manner (0.01–0.1 µM, 2-hour treatment). It also suppressed phosphorylation of downstream signaling proteins PLCγ1, ERK, and Akt in these cells. [1] CH7057288 selectively inhibited proliferation of TRK fusion-positive cell lines (CUTO-3, KM12-Luc, MO-91) with IC₅₀ values in the single- to double-digit nanomolar range (e.g., 9.0 ± 1.9 nM for CUTO-3, 4.4 ± 0.31 nM for KM12-Luc, 1.4 ± 2.0 nM for MO-91). In contrast, TRK-negative cell lines (NCI-H1299, HCT116) were insensitive (IC₅₀ >1,000 nM). In a large-scale screening panel of 272 cancer cell lines, TRK fusion-positive lines were highly enriched among the most sensitive to CH7057288. [1] CH7057288 inhibited wild-type (WT) TRKA and the clinically reported G667C-TRKA mutant with comparable potency (biochemical IC₅₀: WT = 0.0015 µM, G667C = 0.00068 µM; cellular IC₅₀: WT = 0.0047 µM, G667C = 0.0018 µM). However, its activity against the G595R-TRKA mutant was reduced (biochemical IC₅₀ = 0.083 µM; cellular IC₅₀ = 3.0 µM). [1] Gene expression analysis (RNA-seq) of CUTO-3 and KM12-Luc cells treated with 1 µM CH7057288 for 4 and 24 hours revealed significant suppression of the MAPK and E2F pathways downstream of TRK fusion. Western blot analysis confirmed downregulation of CyclinD1, E2F1, p-Rb, and MAPK target genes (DUSP6, SPRY4, ETV1). [1] |
| ln Vivo |
Subcutaneously implanted xenograft tumor models of TRK fusion-positive cells show strong in vivo inhibitory effects on tumor growth. Additionally, CH7057288 significantly induces tumor regression and enhances event-free survival in an intracranial implantation model that mimics brain metastasis. In all three models, CH7057288 induces strong tumor growth inhibition, with notable tumor regression in CUTO-3 and MO-91. Exposure to CH7057288 varies with dose. The plasma concentration decreased to about a few tenths to a hundredth of Tmax 24 hours after the dose due to the relatively short terminal half-life (3 to 5 hours)[1]. CH7057288 induced potent tumor growth inhibition and significant tumor regression in subcutaneous xenograft models of TRK fusion-positive cells (CUTO-3, KM12-Luc, MO-91) upon daily oral administration (0.3–30 mg/kg for 10–28 days). No significant body weight loss was observed at efficacious doses. [1] In an intracranial implantation model using CUTO-3-Luc cells (mimicking CNS metastasis), daily oral administration of CH7057288 (30 mg/kg for 30 days) significantly reduced intracranial bioluminescence signal (indicating tumor regression) and prolonged event-free survival compared to the vehicle control group. [1] In subcutaneous NIH3T3 xenograft models expressing MPRIP-WT-TRKA or MPRIP-G667C-TRKA fusions, CH7057288 (30 mg/kg, once daily for 7 days) induced similar degrees of tumor regression against both WT and G667C mutant tumors. In contrast, tumors expressing the G595R-TRKA mutant were resistant to CH7057288 as well as to entrectinib and larotrectinib. [1] |
| Enzyme Assay |
The inhibitory activity of CH7057288 against TRKA, TRKB, TRKC, and a panel of other kinases (e.g., INSR, ALK, EGFR, FGFR2, HER2, JAK2, KDR, MET, ROS1, SRC, AKT1, CDK1, CHK1, ERK1, PKA, PKCα) was determined using a homogeneous time-resolved fluorescence (HTRF) assay. For tyrosine kinases, enzyme activity towards substrate peptides was measured using a europium-labeled anti-phosphotyrosine antibody. For serine/threonine kinases, activity was measured using a fluorescence polarization (FP) assay. Enzyme-inhibitory activity was calculated, and IC₅₀ values were determined. [1] Kinase selectivity was further assessed using a competitive binding assay (KINOMEscan) against 403 non-mutant and 65 mutant kinases. At 100 nM, CH7057288 bound to only 6 kinases other than TRKA, TRKB, and TRKC. [1] |
| Cell Assay |
The indicated concentrations of CH7057288 are applied for two hours to the TRK fusion-positive cancer cell lines CUTO-3, KM12-Luc, and MO-91. Western blotting is performed after the cells have been lysed. For cell viability/proliferation assays, cells were seeded in spheroid plates or 384-well microplates and treated with serially diluted CH7057288 for 4 or 7 days. Viable cell numbers were quantified using a luminescent ATP detection assay. IC₅₀ values were calculated. [1] For signaling inhibition studies, TRK fusion-positive cell lines were treated with CH7057288 (e.g., 0.01–0.1 µM for 2 hours). Cells were lysed, and phosphorylation levels of TRK, PLCγ1, ERK, and Akt, as well as expression levels of downstream proteins (CyclinD1, E2F1, Rb, DUSP6, SPRY4, ETV1), were analyzed by Western blotting. [1] For mutant TRK activity, NIH3T3 cells stably expressing MPRIP-WT-, G667C-, or G595R-TRKA fusions (established via lentiviral infection and puromycin selection) were treated with CH7057288 and analyzed for cell viability (as above) and TRK phosphorylation (Western blot). [1] For RNA sequencing analysis, CUTO-3 and KM12-Luc cells were treated with 1 µM CH7057288 or DMSO for 4 and 24 hours. Total RNA was extracted, and mRNA libraries were prepared and sequenced. Differentially expressed genes were identified and analyzed for pathway enrichment. [1] |
| Animal Protocol |
For subcutaneous xenograft models, female SCID mice (for MO-91 model) or BALB/c nude mice (for other models) were used. TRK fusion-positive cancer cells (e.g., CUTO-3, KM12-Luc, MO-91) were suspended in HBS or PBS, mixed with Matrigel (for some models), and injected subcutaneously into the right flank. When tumor volumes reached approximately 200–300 mm³, mice were randomized into groups (n=5 or 7). CH7057288, suspended in an unspecified vehicle, was administered orally once daily at doses ranging from 0.3 to 30 mg/kg for 10 to 28 days. Tumor dimensions were measured twice weekly, and volume was calculated (length × width² / 2). For pharmacodynamic analysis, tumors were resected at specified times post-dose for Western blot or RNA analysis. [1] For the intracranial tumor model, CUTO-3-Luc cells were transplanted intracranially into nude mice. Seventeen days post-transplantation, mice were randomized into two groups (n=7) based on intracranial bioluminescence. CH7057288 (30 mg/kg) or vehicle was administered orally once daily for 30 days. Intracranial tumor burden was monitored via bioluminescence imaging after intraperitoneal injection of luciferin. Event-free survival (events defined as death, >20% body weight loss, or motility defect) was recorded. [1] For mutant TRK xenograft studies, NIH3T3 cells expressing MPRIP-WT-, G667C-, or G595R-TRKA fusions were injected subcutaneously into mice. When tumors were established, mice were treated orally once daily for 7 days with CH7057288 (30 mg/kg), entrectinib (150 mg/kg), or larotrectinib (60 mg/kg). Tumor volumes were monitored. [1] |
| ADME/Pharmacokinetics |
Plasma pharmacokinetics of CH7057288 were assessed in tumor-bearing mice after multiple dosing (11 days). Plasma samples were collected pre-dose and at 0.5, 2, 4, 7, and 24 hours post-dose. CH7057288 exhibited dose-dependent plasma exposure. The terminal half-life was relatively short (3–5 hours), with plasma concentration at 24 hours post-dose dropping to approximately a few tenths to a hundredth of the Cmax. At 4 hours post-dose, a 1 mg/kg dose achieved sub-micromolar plasma concentrations, which correlated with strong TRK suppression in tumors. [1] CH7057288 is a potential substrate of P-glycoprotein (P-gp) based on a Caco-2 permeability assay, which may influence its blood-brain barrier penetration. [1] |
| Toxicity/Toxicokinetics |
In the in vivo efficacy studies conducted, daily oral administration of CH7057288 at efficacious doses (up to 30 mg/kg for 10-30 days) did not cause significant body weight loss in mice bearing subcutaneous or intracranial xenografts. [1] In the KM12-Luc subcutaneous model, body weight steadily decreased in the vehicle-treated group but recovered in the CH7057288-treated groups concomitant with tumor growth inhibition. [1] Histological analysis of tumors from CH7057288-treated mice showed decreased Ki-67 (proliferation marker) staining and increased TUNEL (apoptosis marker) staining. [1] |
| References |
[1]. Mol Cancer Ther (2018) 17 (12): 2519–2529. |
| Additional Infomation |
CH7057288 is a novel, selective pan-TRK inhibitor with a benzofuran chemical structure distinct from other reported TRK inhibitors (e.g., entrectinib, larotrectinib). [1] An X-ray crystal structure of the TRKA kinase domain in complex with CH7057288 (PDB ID: 5WR7) revealed that the compound binds in the DFG-out/αC-helix out conformation, forming hydrogen bonds with Met592 and Lys544. This binding mode allows a favorable sulfur-π interaction with the Cys667 residue in the G667C-TRKA mutant, explaining its retained potency against this mutant. In contrast, the G595R mutation causes steric repulsion. [1] CH7057288 suppresses oncogenic signaling downstream of TRK fusions, primarily inhibiting the MAPK pathway initially, followed by suppression of the E2F pathway, leading to cell cycle arrest and apoptosis. [1] The compound demonstrated activity against a clinically reported TRKA resistance mutation (G667C) that confers resistance to entrectinib and larotrectinib, highlighting its potential therapeutic utility in overcoming such resistance. [1] CH7057288 showed efficacy in an intracranial tumor model, suggesting potential utility against TRK fusion-positive cancers with central nervous system involvement. [1] |
Solubility Data
| Solubility (In Vitro) | DMSO: ~100 mg/mL (~175.5 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.75 mg/mL (4.83 mM) (saturation unknown) in 10% DMSO + 40% PEG300 +5% Tween-80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 27.5 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 + to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.7554 mL | 8.7770 mL | 17.5540 mL | |
| 5 mM | 0.3511 mL | 1.7554 mL | 3.5108 mL | |
| 10 mM | 0.1755 mL | 0.8777 mL | 1.7554 mL |