Avapritinib (formerly known as BLU-285; trade name Ayvakit) is an oral, potent and selective small molecule inhibitor of PDGFRα D842V and KIT Exon 17 mutants (IC50=0.5 nM) with anticancer activity. In 2020, it received approval to treat gastrointestinal stromal tumors (GIST) that have spread. In order to treat systemic mastocytosis (SM), a disorder of the mast cells where a KIT Exon 17 mutation is the main cause of disease, avapritinib was being developed as a highly targeted therapy. Patients with advanced systemic mastocytosis saw quick and long-lasting disease control in a phase I trial. Of the patients, 56% had a complete or partial response, for an overall response rate of 72%. Adverse events were mostly mild to moderate in nature, and none of the patients stopped their treatment because of them. Furthermore, BLU-285 might be a useful treatment choice for AMLs with t(8;21) mutations and KIT exon 17 mutations in CBF-AMLs.
Physicochemical Properties
| Molecular Formula | C26H27FN10 | |
| Molecular Weight | 498.5580 | |
| Exact Mass | 498.24 | |
| Elemental Analysis | C, 62.64; H, 5.46; F, 3.81; N, 28.09 | |
| CAS # | 1703793-34-3 | |
| Related CAS # |
|
|
| PubChem CID | 118023034 | |
| Appearance | White to off-white solid powder | |
| LogP | 1.9 | |
| Hydrogen Bond Donor Count | 1 | |
| Hydrogen Bond Acceptor Count | 9 | |
| Rotatable Bond Count | 5 | |
| Heavy Atom Count | 37 | |
| Complexity | 752 | |
| Defined Atom Stereocenter Count | 1 | |
| SMILES | C[C@](C1=CC=C(C=C1)F)(C2=CN=C(N=C2)N3CCN(CC3)C4=NC=NN5C4=CC(=C5)C6=CN(N=C6)C)N |
|
| InChi Key | DWYRIWUZIJHQKQ-SANMLTNESA-N | |
| InChi Code | InChI=1S/C26H27FN10/c1-26(28,20-3-5-22(27)6-4-20)21-13-29-25(30-14-21)36-9-7-35(8-10-36)24-23-11-18(16-37(23)33-17-31-24)19-12-32-34(2)15-19/h3-6,11-17H,7-10,28H2,1-2H3/t26-/m0/s1 | |
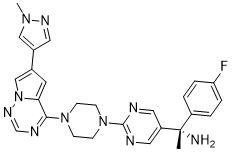
| Chemical Name | (1S)-1-(4-fluorophenyl)-1-[2-[4-[6-(1-methylpyrazol-4-yl)pyrrolo[2,1-f][1,2,4]triazin-4-yl]piperazin-1-yl]pyrimidin-5-yl]ethanamine | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
KIT D816V (IC50 = 0.27 nM); PDGFRA D842V (IC50 = 0.24 nM)
Avapritinib (Blu-285) targets human KIT (wild-type IC50 = 25 nM; exon 11 mutation IC50 = 1.2 nM; exon 17 mutation IC50 = 0.5 nM) and PDGFRα (wild-type IC50 = 39 nM; D842V mutation IC50 = 0.6 nM) [2][3] Avapritinib (Blu-285) exhibits >100-fold selectivity over other kinases including VEGFR2 (IC50 = 4.2 μM), EGFR (IC50 = 5.8 μM), and MET (IC50 = 6.1 μM) [2][3] |
| ln Vitro |
Avapritinib (BLU-285) has shown biochemical in vitro activity on the KIT D816V (IC50=0.27 nM) mutant enzyme, which is a KIT exon 17. Autophosphorylation in the human mast cell leukemia cell line HMC1.2 and the mouse mastocytoma cell line P815, with IC50 values of 4 and 22 nM, respectively, is used to assess the cellular activity of avapritinib on KIT D816 mutants. Avapritinib potently inhibits cellular proliferation (IC50=75 nM), downstream signaling, and KIT N822K mutant autophosphorylation (IC50=40 nM) in Kasumi-1 cells, a t(8;21)-positive AML cell line with a KIT exon 17 N822K mutation[3]. In recombinant kinase activity assays, Avapritinib (Blu-285) dose-dependently inhibited KIT (wild-type and mutants) and PDGFRα (wild-type and D842V) with IC50 values as specified above, acting as an ATP-competitive inhibitor [2][3] - In KIT-mutant gastrointestinal stromal tumor (GIST) cell lines: GIST-T1 (exon 11), GIST48 (exon 11), and GIST430 (exon 17), Avapritinib (Blu-285) inhibited cell proliferation with IC50 values of 0.8 nM, 1.5 nM, and 0.3 nM, respectively (72-hour MTT assay) [2][3] - In PDGFRα D842V-mutant acute myeloid leukemia (AML) cell lines (MV4-11, MOLM-13), Avapritinib (Blu-285) suppressed proliferation with IC50 values of 0.7 nM and 0.9 nM, respectively [2] - Avapritinib (Blu-285) (1-10 nM) reversed ABCB1 and ABCG2-mediated multidrug resistance (MDR) in GIST-T1/MDR and MCF-7/ADR cells: at 5 nM, it restored sensitivity to imatinib (IC50 from >10 μM to 0.6 μM) and doxorubicin (IC50 from >20 μM to 1.8 μM), respectively [1] - Western blot analysis showed that Avapritinib (Blu-285) (5 nM) inhibited KIT/PDGFRα phosphorylation by ~90-95% in respective mutant cell lines, reducing downstream AKT (p-AKT: ~80% inhibition) and STAT5 (p-STAT5: ~85% inhibition) phosphorylation [2][3] - In Annexin V-FITC/PI staining assays, Avapritinib (Blu-285) (5 nM) induced apoptosis in GIST430 cells (35.2% apoptotic rate vs. 4.1% vehicle) and MV4-11 cells (32.6% apoptotic rate vs. 3.8% vehicle) [2][3] - Avapritinib (Blu-285) (up to 1 μM) did not affect the viability of normal human dermal fibroblasts or peripheral blood mononuclear cells (PBMCs) [2][3] |
| ln Vivo |
Avapritinib (BLU-285) exhibits dose-dependent antitumor efficacy and is well tolerated in vivo. With 10 mg/kg once daily oral dosing of Avapritinib, complete inhibition of tumor growth and ≥75% inhibition of KIT kinase is observed in the aggressive KIT exon 17 mutant driven P815 mastocytoma model grown as a solid tumor allograft and in a disseminated model of disease. Throughout the course of the 24-day dosage period, the disease burden in the vehicle control animals increases 86-fold, with widespread disease visible in the femurs, pelvis, and circulating in peripheral blood. This is measured by whole body luciferase imaging (photons/second/mm2). Throughout the course of the trial, avapritinib, at both oral dosages of 10 or 30 mg/kg once daily, significantly lowers the burden of disease. At either 10 or 30 mg/kg, avapritinib causes tumor regression in all animals, and by the end of the study, disease abrogation in multiple animals is indistinguishable from background signal measurements. This in vivo model shows that avapritinib is also well tolerated, and it has no negative effects on body weight at any dose[3]. In nude mice bearing GIST430 (KIT exon 17) xenografts, oral administration of Avapritinib (Blu-285) (10 mg/kg/day or 30 mg/kg/day for 21 days) dose-dependently inhibited tumor growth: high-dose treatment resulted in a tumor growth inhibition (TGI) rate of 82% and reduced tumor weight from 1.45 ± 0.22 g (vehicle) to 0.26 ± 0.05 g [3] - In PDGFRα D842V-mutant AML xenograft mice (MV4-11), oral Avapritinib (Blu-285) (30 mg/kg/day for 14 days) achieved a TGI rate of 75% and prolonged median survival from 22 days (vehicle) to 38 days [2] - Avapritinib (Blu-285) (30 mg/kg/day for 21 days) reversed ABCB1-mediated MDR in GIST-T1/MDR xenograft mice: combination with imatinib (50 mg/kg/day) reduced tumor volume by ~78% compared to imatinib alone (~25% TGI) [1] - Immunohistochemical staining of tumor tissues showed that Avapritinib (Blu-285) (30 mg/kg) reduced p-KIT/p-PDGFRα expression by ~90% and Ki-67 (cell proliferation marker) positivity by ~65% [2][3] |
| Enzyme Assay |
Biochemical in vitro activity of avapritinib (BLU-285) on the KIT exon 17 mutant enzyme, KIT D816V (IC50=0.27 nM), has been demonstrated. LanthaScreen binding assays[2] Serially diluted compounds, KIT protein, and anti–glutathione S-transferase–Europium antibody were incubated with tracer as follows: KIT WT, V560G, and D820E proteins with tracer 222 and KIT D816E, A829P, and D816V with Tracer 178. Plates were incubated at 25°C for 60 min and read on a PerkinElmer EnVision with two excitation flashes and two emission reads: λex = 350 nm, λem = 665 nm; λex = 350 nm, λem = 615 nm.[2] KINOMEscan binding assays[2] Compounds were screened at 3 μM concentration against a panel of 392 WT kinase constructs using the KINOMEscan assay platform at DiscoveRx, as previously described. Hotspot kinase profiling assays[2] In vitro kinase profiling of KIT and PDGFRA constructs was performed at Reaction Biology Corporation. Kinase/substrate pairs, serially diluted compounds, and any additional cofactors required were prepared in a reaction buffer at their respective Michaelis constant (Km) for ATP. 33P-ATP (10 mCi/ml) was added to initiate the reaction, followed by the detection of kinase activity by a filter-binding method. KIT exon 17 mutants conferring ligand-independent constitutive kinase activity are known drivers of disease in both solid tumors and hematologic malignancies. KIT Exon 17 mutations have been identified in several human diseases such as systemic mastocytosis (SM), as resistance mutations in second and third line gastrointestinal stromal tumor (GIST), and certain subsets of acute myeloid leukemia (AML). We have developed BLU-285, a potent and selective exon 17 mutant KIT kinase inhibitor. BLU-285 has demonstrated biochemical in vitro activity on the KIT exon 17 mutant enzyme, KIT D816V (half-maximal inhibitory concentration [IC50] = 0.27 nM). Cellular activity of BLU-285 on KIT D816 mutants was measured by autophosphorylation in the human mast cell leukemia cell line HMC1.2, and the P815 mouse mastocytoma cell line with IC50= 4 and 22 nM, respectively.[3] KIT/PDGFRα kinase activity assay: Recombinant human KIT (wild-type, exon 11, exon 17 mutants) and PDGFRα (wild-type, D842V mutant) kinase domains were individually incubated with reaction buffer containing ATP (10 μM) and a fluorescent peptide substrate. Serial dilutions of Avapritinib (Blu-285) (0.001-1000 nM) were added, and the mixture was incubated at 37°C for 45 minutes. The reaction was terminated by adding EDTA-containing stop solution, and fluorescence intensity (excitation 485 nm, emission 535 nm) was measured to assess peptide phosphorylation. IC50 values were calculated by nonlinear regression of dose-response curves [2][3] - Kinase selectivity assay: A panel of 40 recombinant kinases (including VEGFR2, EGFR, MET) was subjected to the same kinase assay protocol. Avapritinib (Blu-285) (0.001-10 μM) was tested to determine IC50 values for these kinases, confirming selective inhibition of KIT and PDGFRα [2][3] |
| Cell Assay |
Autophosphorylation in the human mast cell leukemia cell line HMC1.2 and the mouse mastocytoma cell line P815, with IC50 values of 4 and 22 nM, respectively, is used to assess the cellular activity of avapritinib on KIT D816 mutants. Avapritinib potently inhibits cellular proliferation (IC50=75 nM), downstream signaling, and KIT N822K mutant autophosphorylation (IC50=40 nM) in Kasumi-1 cells, a t(8;21)-positive AML cell line with a KIT exon 17 N822K mutation. GIST/AML cell proliferation assay: GIST-T1, GIST48, GIST430, MV4-11, and MOLM-13 cells were seeded in 96-well plates at 5×10³ cells/well. After 24-hour attachment, serial dilutions of Avapritinib (Blu-285) (0.01-100 nM) were added, and cells were cultured for 72 hours. MTT reagent was added, and absorbance at 570 nm was measured to calculate cell viability and IC50 values [2][3] - Multidrug resistance reversal assay: GIST-T1/MDR and MCF-7/ADR cells were seeded in 96-well plates (5×10³ cells/well) and pre-treated with Avapritinib (Blu-285) (1-10 nM) for 1 hour, then co-treated with imatinib (0.1-10 μM) or doxorubicin (0.1-20 μM) for 72 hours. Cell viability was assessed by MTT assay to determine reversal of MDR [1] - Apoptosis assay: GIST430 and MV4-11 cells were seeded in 6-well plates (2×10⁵ cells/well) and treated with Avapritinib (Blu-285) (1-10 nM) for 48 hours. Cells were stained with Annexin V-FITC and PI, then analyzed by flow cytometry to determine apoptotic rate [2][3] - Western blot assay: KIT/PDGFRα-mutant cells were treated with Avapritinib (Blu-285) (0.5-50 nM) for 24 hours. Cells were lysed in RIPA buffer, and proteins were probed with antibodies against p-KIT, KIT, p-PDGFRα, PDGFRα, p-AKT, AKT, p-STAT5, STAT5, and GAPDH (loading control) [2][3] |
| Animal Protocol |
Mice: The effectiveness of avapritinib (BLU-285) in KIT exon 17-mutated CBF-AML is evaluated in a femoral injection model of Kasumi-1 luc+ AML NOG SCID mice. Mice are dosed with avapritinib orally once daily at 10 mg/kg or 30 mg/kg through day 45 after a 21-day post-injection latency period. All animal studies were performed under IACUC guidelines established at each respective institution where the study was conducted. Avapritinib (BLU-28) was formulated in 0.5% carboxymethyl cellulose + 1% Tween 80. Dasatinib was formulated in 50% propylene glycol. Imatinib was formulated in sterile water. Regorafenib was formulated in a PEG400/125 mM methanesulfonic acid (MSA) mixture at 80:20 ratio. MSA (125 mM) was prepared in water. Sunitinib was formulated in 50 mM citrate buffer (pH 3.5). The P815 xenograft study was performed at WuXi AppTec in Shanghai, China. BALB/c nude mice were inoculated with 1 × 106 P815 cells subcutaneously at the right flank. When the average tumor size reached about 75 mm3, treatment with test article began. Ten animals were treated per group. Tumors were measured to assess antitumor activity. For PK/PD analysis, plasma and tumors were collected from three mice per group. The GIST exon 11/17 (model 2007031011) and exon 11/13 (model GS11331) xenograft studies were performed at Crown Biosciences. Nonobese diabetic–severe combined immunodeficient mice were inoculated with 100,000 to 125,000 viable cells subcutaneously into the rear flank. Animals were randomized when average tumor volume reached 150 to 200 mm3, followed by oral dosing of compounds. Eleven animals were treated per group. Plasma and tumor samples were collected from three animals per group. [2] GIST430 xenograft model: Female BALB/c nude mice (4-6 weeks old) were subcutaneously implanted with 5×10⁶ GIST430 cells. When tumors reached ~100 mm³, mice were randomly divided into vehicle control, Avapritinib (Blu-285) 10 mg/kg, and 30 mg/kg groups (n=6 per group). The drug was dissolved in 0.5% methylcellulose + 0.2% Tween 80 and administered by oral gavage once daily for 21 days. Tumor volume was measured every 3 days, and tumor weight was recorded at the end of treatment [3] - MV4-11 AML xenograft model: Female nude mice (4-6 weeks old) were subcutaneously implanted with 5×10⁶ MV4-11 cells. When tumors reached ~150 mm³, mice were assigned to vehicle or Avapritinib (Blu-285) 30 mg/kg groups (n=7 per group). The drug was formulated as described above and administered orally once daily for 14 days. Survival was monitored for 45 days, and tumor tissues were collected for immunohistochemical staining [2] - GIST-T1/MDR MDR reversal model: Nude mice bearing GIST-T1/MDR xenografts were randomly divided into four groups (n=6 per group): vehicle, imatinib alone (50 mg/kg/day), Avapritinib (Blu-285) alone (30 mg/kg/day), and combination. Drugs were administered orally once daily for 21 days. Tumor volume was measured every 3 days to assess MDR reversal efficacy [1] |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion A 300mg oral dose of avapritinib reaches a Cmax of 813ng/mL with a Tmax of 2.0-4.1h and an AUC of 15400h\*ng/mL. Avapritinib is 70% eliminated in the feces with 11% as the unchanged drug and 18% eliminated in the urine with 0.23% as the unchanged drug. The mean apparent volume of distribution is 1200L. The mean apparent oral clearance of avapritinib is 19.5L/h. Metabolism / Metabolites Avapritinib is metabolized mainly by CYP3A4 and CYP2C9 _in vitro_. A 310mg oral dose is recovered as 49% unchanged drug, 35% hydroxy glucuronide metabolite, and 14% oxidatively deaminated metabolite. Biological Half-Life The half life of avapritinib is 32-57h. Oral bioavailability: In mice, oral administration of Avapritinib (Blu-285) (30 mg/kg) resulted in an oral bioavailability of ~52% [2] - Plasma half-life (t1/2): In mice, t1/2 = 6.8 ± 0.9 hours (oral 30 mg/kg); in rats, t1/2 = 8.5 ± 1.2 hours (oral 20 mg/kg); in humans, t1/2 = 32.1 ± 4.5 hours (oral 300 mg) [2] - Peak plasma concentration (Cmax): In mice, oral 30 mg/kg achieved Cmax = 412 ± 53 ng/mL at 1.8 ± 0.3 hours post-dosing; in humans, oral 300 mg achieved Cmax = 896 ± 124 ng/mL at 4.0 ± 0.5 hours [2] - AUC0-∞: In mice, AUC0-∞ = 3280 ± 410 ng·h/mL (oral 30 mg/kg); in humans, AUC0-∞ = 28,500 ± 3600 ng·h/mL (oral 300 mg) [2] - Volume of distribution (Vd/F): In humans, Vd/F = 16.2 ± 2.3 L (oral 300 mg) [2] - Clearance (CL/F): In humans, CL/F = 10.5 ± 1.4 mL/min (oral 300 mg) [2] - Metabolism: Avapritinib (Blu-285) is primarily metabolized by CYP3A4 in human liver microsomes, with minor contributions from CYP2C9 and CYP2C19 [2] |
| Toxicity/Toxicokinetics |
Hepatotoxicity In the prelicensure clinical trials of avapritinib in patients with GIST harboring the PDGFRA D842V mutation, ALT elevations arose in 20% of patients but were usually self-limited and mild. ALT elevations above 5 times the upper limit of normal (ULN) were uncommon, being found in 1% to 3% of treated patients. In the open label trials supporting the approval of avapritinib, there were no instances of clinically apparent liver injury, hepatic failure or deaths from liver injury. Avapritinib is associated with frequent elevations in serum bilirubin, but the increases are largely indirect and resolve rapidly with discontinuation. Likelihood score: E* (unproven but suspected rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the clinical use of avapritinib during breastfeeding. Because avapritinib is 98.8% bound to plasma proteins, the amounts in milk are likely to be low. However, the manufacturer recommends avoiding breastfeeding during therapy and for 2 weeks following the final dose. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Avapritinib is 98.8% protein bound in serum. In vitro cytotoxicity: Avapritinib (Blu-285) exhibited CC50 > 1 μM in normal human dermal fibroblasts and PBMCs [2][3] - Acute toxicity in mice: Single oral administration of Avapritinib (Blu-285) up to 500 mg/kg did not cause mortality or overt toxicity (lethargy, weight loss) [2] - Chronic toxicity in rats: Repeated oral administration of Avapritinib (Blu-285) (30 mg/kg/day for 28 days) did not induce significant changes in hematological parameters (RBC, WBC, platelets) or serum biochemical markers (ALT, AST, creatinine, BUN) [2] - Plasma protein binding: Avapritinib (Blu-285) exhibited plasma protein binding of 97-99% in mouse, rat, and human plasma (equilibrium dialysis) [2][3] - Drug-drug interaction: Avapritinib (Blu-285) is a weak inhibitor of CYP3A4 (IC50 = 8.7 μM) and does not inhibit other major CYP450 isoforms [2] |
| References |
[1]. Avapritinib: A Selective Inhibitor of KIT and PDGFRα that Reverses ABCB1 and ABCG2-MediatedMultidrug Resistance in Cancer Cell Lines. Mol Pharm. 2019 Jul 1;16(7):3040-3052. [2]. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci Transl Med. 2017 Nov 1;9(414). pii: eaao1690. [3]. Blu-285, a Potent and Selective Inhibitor for Hematologic Malignancies with KIT Exon 17 Mutations.Blood 2015 126:568. |
| Additional Infomation |
Avapritinib, or BLU-285, is a selective tyrosine kinase inhibitor of KIT and platelet derived growth factor receptor alpha indicated for the treatment of unresectable, metastatic gastrointestinal stromal tumors and advanced systemic mastocytosis. It is one of the first medications available for the treatment of multidrug resistant cancers. Avapritinib shares a similar mechanism with [ripretinib]. Avapritinib was granted FDA approval on 9 January 2020 and EMA approval on 24 September 2020. Avapritinib is a Kinase Inhibitor. The mechanism of action of avapritinib is as a Tyrosine Kinase Inhibitor, and Cytochrome P450 2C9 Inhibitor, and P-Glycoprotein Inhibitor, and Breast Cancer Resistance Protein Inhibitor, and Multidrug and Toxin Extrusion Transporter 1 Inhibitor, and Multidrug and Toxin Extrusion Transporter 2 K Inhibitor, and Bile Salt Export Pump Inhibitor. Avapritinib is a tyrosine kinase receptor inhibitor that targets mutant forms of several genes (KIT, PDGFRA) involved in gastrointestinal stromal tumors which are often found in refractory cases. Serum aminotransferase elevations arise in a small proportion of patients treated with the highest doses of avapritinib, but episodes of clinically apparent liver injury have not been reported with its use. Avapritinib is an orally bioavailable inhibitor of specific mutated forms of platelet-derived growth factor receptor alpha (PDGFR alpha; PDGFRa) and mast/stem cell factor receptor c-Kit (SCFR), with potential antineoplastic activity. Upon oral administration, avapritinib specifically binds to and inhibits specific mutant forms of PDGFRa and c-Kit, including the PDGFRa D842V mutant and various KIT exon 17 mutants. This results in the inhibition of PDGFRa- and c-Kit-mediated signal transduction pathways and the inhibition of proliferation in tumor cells that express these PDGFRa and c-Kit mutants. PDGFRa and c-Kit, protein tyrosine kinases and tumor-associated antigens (TAAs), are mutated in various tumor cell types; they play key roles in the regulation of cellular proliferation. Drug Indication Avapritinib is indicated for the treatment of adults with unresectable or metastatic GIST harboring a platelet-derived growth factor receptor alpha (PDGFRA) exon 18 mutation, including PDGFRA D842V mutations. It is also used to treat adult patients with advanced systemic mastocytosis (AdvSM). AdvSM includes patients with aggressive systemic mastocytosis (ASM), systemic mastocytosis with an associated hematological neoplasm (SM-AHN), and mast cell leukemia. However, it is not recommended for the treatment of patients with AdvSM with platelet counts of less than 50 X 109 L. Ayvakyt is indicated as monotherapy for the treatment of adult patients with unresectable or metastatic gastrointestinal stromal tumours (GIST) harbouring the platelet-derived growth factor receptor alpha (PDGFRA) D842V mutation. Treatment of all conditions included in the category of malignant neoplasms (except haematopoietic and lymphoid tissue neoplasms) Treatment of mastocytosis Mechanism of Action Avapritinib has a negative modulating effect on the transporters ABCB1 and ABCG2, which mediate the multidrug resistance phenotype of some cancers. This modulation may be due to interactions of avapritinib with the drug binding pocket of these transporters. Negative modulation of these transporters, resensitizes cancerous cells to treatment with chemotherapeutic agents like [paclitaxel]. Avapritinib (Blu-285) is a potent, orally active, and highly selective small-molecule inhibitor of KIT and PDGFRα kinases, with specific activity against clinically relevant mutations (e.g., KIT exon 17, PDGFRα D842V) [2][3] - The therapeutic mechanism involves ATP-competitive inhibition of KIT/PDGFRα, blocking downstream proliferative and survival signaling pathways (AKT/STAT5), and reversing ABCB1/ABCG2-mediated multidrug resistance by inhibiting transporter function [1][2][3] - Avapritinib (Blu-285) was approved by the FDA for the treatment of unresectable or metastatic GIST harboring KIT exon 17 mutations and PDGFRα D842V-mutant AML [2] - Preclinical and clinical data demonstrate significant efficacy in KIT/PDGFRα-mutant cancers, with a favorable safety profile and minimal off-target effects [1][2][3] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.01 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.01 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.01 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 2% DMSO+40% PEG 300+2% Tween 80+ddH2O: 4mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0058 mL | 10.0289 mL | 20.0578 mL | |
| 5 mM | 0.4012 mL | 2.0058 mL | 4.0116 mL | |
| 10 mM | 0.2006 mL | 1.0029 mL | 2.0058 mL |