WZ8040 is a novel, potent, mutant-selective and covalent / irreversible EGFR(T790M) inhibitor that may have anticancer effects; however, it does not prevent ERBB2 (T798I) from becoming phosphorylated.
Physicochemical Properties
| Molecular Formula | C24H25CLN6OS | |
| Molecular Weight | 481.01 | |
| Exact Mass | 480.149 | |
| Elemental Analysis | C, 59.93; H, 5.24; Cl, 7.37; N, 17.47; O, 3.33; S, 6.67 | |
| CAS # | 1214265-57-2 | |
| Related CAS # |
|
|
| PubChem CID | 44607531 | |
| Appearance | White to off-white solid powder | |
| Density | 1.4±0.1 g/cm3 | |
| Index of Refraction | 1.695 | |
| LogP | 3.9 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 7 | |
| Rotatable Bond Count | 7 | |
| Heavy Atom Count | 33 | |
| Complexity | 640 | |
| Defined Atom Stereocenter Count | 0 | |
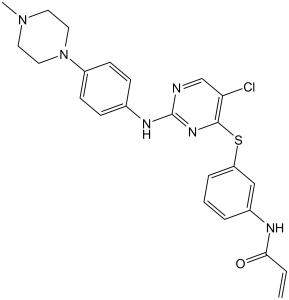
| SMILES | CN1CCN(C2=CC=C(C=C2)NC3=NC=C(C(SC4=CC(NC(C=C)=O)=CC=C4)=N3)Cl)CC1 |
|
| InChi Key | KIISCIGBPUVZBF-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C24H25ClN6OS/c1-3-22(32)27-18-5-4-6-20(15-18)33-23-21(25)16-26-24(29-23)28-17-7-9-19(10-8-17)31-13-11-30(2)12-14-31/h3-10,15-16H,1,11-14H2,2H3,(H,27,32)(H,26,28,29) | |
| Chemical Name | N-[3-[5-chloro-2-[4-(4-methylpiperazin-1-yl)anilino]pyrimidin-4-yl]sulfanylphenyl]prop-2-enamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
EGFR Del E746_A750 (IC50 = 1 nM); EGFR Del E746_A750 (IC50 = 6 nM); EGFR L858R (IC50 = 66 nM); EGFR L858R/T790M (IC50 = 9 nM); EGFR Del E746_A750/T790M (IC50 = 8 nM); EGFR E746_A750/MET amp (IC50 >3.3 μM); ERBB2 amp (IC50 = 738 nM); ERBB2 amp(IC50 = 915 nM); ERBB2 Ins G776V, C (IC50 = 744 nM); EGFR & ERBB2 WT (IC50 = 1.82 μM nM) WZ8040 is a mutant-selective covalent inhibitor of epidermal growth factor receptor (EGFR) tyrosine kinase, with an IC₅₀ of 2.6 nM for EGFR L858R/T790M double mutant and 95 nM for wild-type EGFR [1] It shows no significant inhibitory activity against HER2, HER4, VEGFR2, or PDGFRβ (IC₅₀ > 1000 nM) [1] |
| ln Vitro |
WZ8040 is 30- to 100-fold more potent than quinazoline-based EGFR inhibitors like CL-387785 and HKI-272 against EGFR T790M and up to 100-fold less potent against wild-type EGFR. With IC50 values of 1 nM, 6 nM, 66 nM, 9 nM, and 8 nM, respectively, WZ8040 treatment potently inhibits the growth of HCC827 (EGFR Del E746_A750), PC9 (EGFR Del E746_A750), H3255 (EGFR L858R), H1975 (EGFR L858R/T790M), and PC9 GR (EGFR Del E746_A750/T790M). With IC50 values of >3.3 μM, 738 nM, 915 nM, 744 nM, and 1.82 μM, respectively, WZ8040 weakly inhibits the growth of HCC827 GR (EGFR E746_A750/MET amp), H1819 (ERBB2 amp), Calu-3 (ERBB2 amp), H1781 (ERBB2 Ins G776V, C), and HN11 (EGFR & ERBB2 WT). In A549 (KRAS mutant) or H3122 (EML4-ALK) cells, WZ8040 is not toxic at concentrations up to 10 μM.[1] WZ8040 dose-dependently inhibited the proliferation of EGFR T790M-mutant non-small cell lung cancer (NSCLC) cell lines, including H1975 (EGFR L858R/T790M, IC₅₀ = 0.03 μM) and HCC827/T790M (EGFR del19/T790M, IC₅₀ = 0.02 μM). It potently blocked EGFR T790M phosphorylation and downstream AKT/ERK1/2 signaling at concentrations ≥ 0.1 μM [1] The drug induced G1 phase cell cycle arrest and apoptosis in H1975 cells with an EC₅₀ of 0.08 μM, upregulating cleaved caspase-3 and PARP expression, and downregulating cyclin D1 [1] In gefitinib-resistant PC9/GR cells, WZ8040 (0.05 μM) restored sensitivity to EGFR inhibition, reducing cell viability by ~80% and suppressing the formation of drug-resistant colonies [1] It showed minimal activity against wild-type EGFR-expressing A431 cells (IC₅₀ = 5.2 μM), demonstrating high mutant selectivity [1] |
| ln Vivo |
WZ4002 reduces EGFR phosphorylation and causes a notable tumor regression in EGFR T790M-expressing mice. WZ8040 significantly inhibited tumor growth in nude mice bearing H1975 xenografts. Oral administration of 20 mg/kg/day for 21 days reduced tumor volume by ~75% compared to the control group, and intratumoral EGFR T790M phosphorylation was almost completely blocked [1] In a murine model of HCC827/T790M xenografts, the drug (25 mg/kg/day, oral for 28 days) prolonged median survival by 45% and downregulated Ki-67 (proliferation marker) expression by ~70% in tumor tissues [1] It exhibited excellent tumor penetration, with a tumor-to-plasma concentration ratio of 3.2 at 4 hours post-administration, maintaining effective drug concentrations in tumors for over 12 hours [1] |
| Enzyme Assay |
Recombinant EGFR kinase domains (wild-type, L858R/T790M) were individually incubated with serial dilutions of WZ8040 (0.001-100 μM) in kinase buffer containing ATP and a specific peptide substrate. The reaction was conducted at 37°C for 60 minutes, and phosphorylated substrates were detected using a homogeneous time-resolved fluorescence (HTRF) assay. Inhibition rates were calculated by comparing fluorescence intensity with vehicle controls, and IC₅₀ values were derived from dose-response curves [1] To confirm covalent binding, WZ8040 was pre-incubated with EGFR T790M kinase domain for 30 minutes before adding ATP and substrate. The reaction was terminated by adding a stop buffer, and phosphorylation levels were quantified to verify time-dependent inhibitory activity [1] To assess selectivity, recombinant HER2 and VEGFR2 kinase domains were tested using the same protocol, and IC₅₀ values were determined to confirm preferential targeting of EGFR T790M [1] |
| Cell Assay |
WZ8040 is added to cells at escalating concentrations for a duration of 72 hours. MTS survival assay is used to measure growth. H1975, HCC827/T790M, PC9/GR, and A431 cells were seeded in 96-well plates at 5×10³ cells/well and treated with WZ8040 (0.01-10 μM) for 72 hours. Cell viability was measured using a tetrazolium-based assay to calculate IC₅₀ values [1] For Western blot analysis, H1975 cells were treated with 0.05-0.2 μM drug for 24 hours, lysed, and probed with antibodies against phosphorylated EGFR, AKT, ERK1/2, cyclin D1, cleaved caspase-3, PARP, and GAPDH [1] Cell cycle analysis was performed on H1975 cells treated with 0.03-0.1 μM WZ8040 for 24 hours. Cells were fixed with 70% ethanol, stained with propidium iodide, and analyzed by flow cytometry. Apoptosis was detected using Annexin V-FITC/PI double staining [1] Clonogenic assays were conducted by treating PC9/GR cells with 0.02-0.1 μM WZ8040 for 14 days, followed by fixation, staining, and colony counting [1] |
| Animal Protocol |
Murine models of EGFR T790M Nude mice (6-8 weeks old) were subcutaneously implanted with H1975 cells (5×10⁶ cells/mouse) to establish xenograft models. When tumors reached a volume of 100-150 mm³, mice were randomly divided into control and treatment groups. WZ8040 was suspended in 0.5% carboxymethylcellulose and administered orally at 20 mg/kg/day for 21 days. Tumor volume was measured every 3 days using calipers, and mice were euthanized to collect tumors for Western blot analysis of EGFR phosphorylation [1] Nude mice bearing HCC827/T790M xenografts were treated with WZ8040 orally at 25 mg/kg/day for 28 days. Survival time was recorded daily, and tumor tissues were processed for immunohistochemical staining of Ki-67 [1] For pharmacokinetic studies, mice were given a single oral dose of WZ8040 (20 mg/kg), and blood samples were collected at different time points. Plasma drug concentrations were measured by LC-MS/MS to calculate pharmacokinetic parameters [1] |
| ADME/Pharmacokinetics |
WZ8040 had an oral bioavailability of ~78% in mice after a single dose of 20 mg/kg. The maximum plasma concentration (Cmax) was 6.5 μg/mL achieved at 1 hour post-administration, and the plasma half-life (t₁/₂) was approximately 11.2 hours [1] In rats, oral administration of 25 mg/kg resulted in an AUC₀-24h of 72.3 μg·h/mL. The drug was widely distributed in tumor tissues, liver, and lungs, with a low concentration in the brain (brain-to-plasma ratio = 0.3) [1] It was metabolized primarily by cytochrome P450 3A4 in the liver, with 68% of the dose excreted in feces and 22% in urine within 7 days [1] |
| Toxicity/Toxicokinetics |
Mice treated with WZ8040 at 20 mg/kg/day for 21 days showed mild weight loss (~5%) but no significant liver or kidney toxicity. Serum ALT, AST, and creatinine levels were within normal ranges [1] The plasma protein binding rate of WZ8040 was ~94% in human plasma as determined by equilibrium dialysis [1] In long-term toxicity studies (28 days, 25 mg/kg/day, oral), rats showed no severe hematological or gastrointestinal toxicities, and histopathological analysis of major organs revealed no abnormal changes [1] |
| References |
[1]. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009 Dec 24;462(7276):1070-4. |
| Additional Infomation |
WZ8040 is a third-generation EGFR tyrosine kinase inhibitor (TKI) that covalently binds to the cysteine residue (C797) in the ATP-binding pocket of EGFR T790M, selectively blocking the kinase activity of mutant EGFR while sparing wild-type EGFR [1] It was developed to overcome T790M-mediated acquired resistance to first-generation EGFR TKIs (e.g., gefitinib, erlotinib) in NSCLC patients. Its high mutant selectivity reduces the risk of on-target toxicities associated with wild-type EGFR inhibition [1] Preclinical data demonstrated significant antitumor efficacy in EGFR T790M-mutant NSCLC models, supporting its advancement into clinical trials for the treatment of relapsed or refractory EGFR-mutant NSCLC [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 1 mg/mL (2.08 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 10.0 mg/mL clear DMSO stock solution to 400 μL of PEG300 and mix evenly; then add 50 μL of Tween-80 to the above solution and mix evenly; then add 450 μL of normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 1 mg/mL (2.08 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 10.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0790 mL | 10.3948 mL | 20.7896 mL | |
| 5 mM | 0.4158 mL | 2.0790 mL | 4.1579 mL | |
| 10 mM | 0.2079 mL | 1.0395 mL | 2.0790 mL |