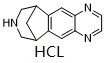
Varenicline dihydrochloride is a nicotinic receptor partial agonist. Varenicline dihydrochloride is a partial agonist on the α4β2, α3β4, and α6β2 subtypes. Varenicline dihydrochloride displays full agonism on α7 nicotinic acetylcholine receptors.
Physicochemical Properties
| Molecular Formula | C13H15CL2N3 |
| Molecular Weight | 284.1843 |
| Exact Mass | 247.088 |
| Elemental Analysis | C, 54.94; H, 5.32; Cl, 24.95; N, 14.79 |
| CAS # | 866823-63-4 |
| Related CAS # | Varenicline;249296-44-4;Varenicline-d4 hydrochloride;Varenicline-d4 dihydrochloride |
| PubChem CID | 45263226 |
| Appearance | Brown to dark brown solid powder |
| LogP | 2.934 |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 3 |
| Rotatable Bond Count | 0 |
| Heavy Atom Count | 18 |
| Complexity | 254 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | Cl.N1C2C(=CC3C4CC(CNC4)C=3C=2)N=CC=1 |
| Synonyms | Varenicline dihydrochloride; HSDB7591; HSDB-7591; HSDB 7591; CP 526555; CP-526555; CP526555; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment (e.g. under nitrogen), avoid exposure to moisture and light. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Varenicline dihydrochloride targets α4β2 neuronal nicotinic acetylcholine receptor (nAChR) as a partial agonist, with a Ki value of 0.14 nM (radioligand binding assay) [2] Varenicline dihydrochloride targets α7 nAChR as a full agonist, with an EC₅₀ value of 1.2 μM (ion channel activation assay) [2] |
| ln Vitro |
RAW 264.7 macrophages' LPS-induced cytokine secretion (IL-1β, IL-6, and TNFα) and cell proliferation rate are inhibited by vannicline diHClide (1 μM, 24 h) [1]. Human adrenal chromaffin cells separated from male and female organ donors exhibit action potentials (Aps) stimulation in the absence of ACh stimulation when exposed to 250 nM vannicline diHClide [3]. By lowering VE-cadherin protein expression, vannicline diHClide (100 μM, 4 h) stimulates HUVEC migration [4]. In LPS-stimulated RAW 264.7 macrophages: Varenicline dihydrochloride (1–10 μM) dose-dependently inhibited inflammatory cytokine production, reducing TNF-α secretion by 35–68% and IL-6 by 40–72% (ELISA); it also suppressed NF-κB p65 nuclear translocation and p38 MAPK phosphorylation (Western blot) [1] - In human umbilical vein endothelial cells (HUVECs): Varenicline dihydrochloride (0.1–5 μM) promoted cell migration by 2.1–3.8-fold (scratch assay), downregulated vascular endothelial-cadherin (VE-cadherin) expression by 45–70% (Western blot), and activated ERK1/2 and p38 MAPK phosphorylation via α7 nAChR [4] - In human adrenal chromaffin cells: Therapeutic concentrations (0.1–1 μM) of Varenicline dihydrochloride in the presence of nicotine (1 μM) increased action potential firing frequency by 2.5-fold and prolonged action potential duration, as measured by patch-clamp electrophysiology [3] - At α4β2 nAChR: Varenicline dihydrochloride (0.01–1 μM) induced partial ion channel activation (40% of maximal nicotine response) in Xenopus oocytes expressing human α4β2 receptors [2] - At α7 nAChR: Varenicline dihydrochloride (0.1–10 μM) induced full ion channel activation (equivalent to nicotine) in oocytes expressing human α7 receptors [2] - No significant cytotoxicity was observed in RAW 264.7, HUVECs, or adrenal chromaffin cells at concentrations up to 20 μM [1][3][4] |
| ln Vivo |
Nicotine conditioned place preference (CPP) is inhibited by vannicline disalk (0.01-1 mg/kg subcutaneously, 3 days), when administered 10 minutes prior to nicotine (0.5 mg/kg subcutaneously) [5]. Position aversion caused by vannicline diHClide (subcutaneous injection, 2.5 mg/kg, 3 days) is dependent on α5 nAChR but not β2 nAChR [5]. Subcutaneous injection of vannicline diHClide (0.1 and 0.5 mg/kg, 3 days) reverses the somatic symptoms and hyperalgesia associated with nicotine withdrawal, as well as withdrawal-induced aversion, in a dose-related manner [5]. In nicotine-sensitized C57BL/6 mice: Oral administration of Varenicline dihydrochloride (1, 3 mg/kg, once daily for 7 days) dose-dependently reduced nicotine-induced conditioned place preference (CPP) score by 32% and 58%, respectively [5] - In nicotine withdrawal mice model: Varenicline dihydrochloride (3 mg/kg po) reduced withdrawal-related behaviors (jumping frequency decreased by 65%) and alleviated nicotine-induced hyperalgesia, increasing thermal withdrawal latency by 42% (hot plate test) [5] - The compound did not induce CPP or aversive effects when administered alone in mice [5] |
| Enzyme Assay |
α4β2 nAChR binding assay: Membranes from cells expressing human α4β2 nAChR were incubated with [³H]-nicotine and serial dilutions of Varenicline dihydrochloride at 25°C for 2 hours. Bound radioligand was separated by filtration, and radioactivity was measured to calculate Ki value [2] - α7 nAChR functional assay: Xenopus oocytes were injected with cRNA encoding human α7 nAChR. After 2–3 days of incubation, Varenicline dihydrochloride was applied, and ion currents were recorded using two-electrode voltage clamp to determine EC₅₀ and agonist efficacy [2] |
| Cell Assay |
Cell proliferation assay [1] Cell Types: RAW 264.7 mouse macrophages (treated with 4 μg/mL LPS for 24 h) Tested Concentrations: 1 μM Incubation Duration: 0-48 h Experimental Results: LPS-induced cell proliferation rate diminished. Western Blot Analysis[4] Cell Types: HUVEC Tested Concentrations: 1, 10, 100 μM Incubation Duration: 24 hrs (hours) or 30 minutes Experimental Results: diminished VE-cadherin protein expression, activation of ERK1/2, p38 and JNK signaling. Macrophage inflammation assay (RAW 264.7): Cells were seeded in 24-well plates, pretreated with Varenicline dihydrochloride (1–10 μM) for 1 hour, then stimulated with LPS (1 μg/mL) for 24 hours. Culture supernatants were collected for TNF-α/IL-6 quantification by ELISA; cell lysates were used for Western blot analysis of NF-κB p65 and p38 MAPK [1] - Endothelial cell migration assay (HUVECs): Cells were cultured to confluence, scratched with a pipette tip, and treated with Varenicline dihydrochloride (0.1–5 μM). Migration distance was measured at 0 and 24 hours using image analysis software; Western blot was performed to detect VE-cadherin and MAPK phosphorylation [4] - Electrophysiology assay (human adrenal chromaffin cells): Cells were isolated and cultured, then treated with Varenicline dihydrochloride (0.1–1 μM) in the presence of nicotine (1 μM). Action potentials were recorded using whole-cell patch-clamp technique [3] |
| Animal Protocol |
Animal/Disease Models: ICR male mice [5] Doses: 0.01-1 mg/kg, 3 days Route of Administration: subcutaneous injection Experimental Results: Inhibited nicotine conditioned place preference (CPP) in a dose-dependent manner. Nicotine CPP mouse model: Male C57BL/6 mice (20–25 g) were sensitized with nicotine (0.5 mg/kg ip) once daily for 7 days. Varenicline dihydrochloride was administered orally (1, 3 mg/kg) 30 minutes before each nicotine injection. CPP was tested in a two-compartment apparatus on day 8 [5] - Nicotine withdrawal and hyperalgesia model: Mice were implanted with nicotine pellets (7.2 mg) for 14 days to induce dependence. Pellets were removed to trigger withdrawal, and Varenicline dihydrochloride (3 mg/kg po) was administered once daily for 3 days. Withdrawal behaviors (jumping) were counted for 30 minutes; thermal hyperalgesia was assessed by hot plate test [5] - Drug formulation: Varenicline dihydrochloride was dissolved in normal saline for oral administration [5] |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Varenicline undergoes minimal metabolism, with 92% excreted unchanged in the urine. Renal elimination of varenicline is primarily through glomerular filtration along with active tubular secretion possibly via the organic cation transporter, OCT2. Maximum plasma concentrations of varenicline occur typically within 3-4 hours after oral administration. Following administration of multiple oral doses of varenicline, steady-state conditions were reached within 4 days. Over the recommended dosing range, varenicline exhibits linear pharmacokinetics after single or repeated doses. In a mass balance study, absorption of varenicline was virtually complete after oral administration and systemic availability was ~90%. Oral bioavailability of varenicline is unaffected by food or time-of-day dosing. Plasma protein binding of varenicline is low ( Varenicline is eliminated principally in urine as unchanged drug. Renal elimination of the drug occurs primarily through glomerular filtration along with active tubular secretion. Varenicline is distributed into milk in animals. Not known whether varenicline is distributed into human milk. Metabolism / Metabolites Metabolism is limited (<10%). Most of the active compound is excreted by the kidneys (81%). A minor amount of varenicline is glucuronidated, oxidated, N-formylated, as well as conjugated to form a hexose. Varenicline undergoes minimal metabolism, with 92% excreted unchanged in the urine. Varenicline undergoes minimal metabolism with 92% excreted unchanged in the urine and less than 10% excreted as metabolites. Minor metabolites in urine include varenicline N-carbamoylglucuronide and hydroxyvarenicline. In circulation, varenicline comprises 91% of drug related material. Minor circulating metabolites include varenicline N-carbamoylglucuronide and N-glucosylvarenicline. Biological Half-Life The elimination half-life of varenicline is approximately 24 hours The elimination half-life of varenicline is approximately 24 hours. |
| Toxicity/Toxicokinetics |
Hepatotoxicity Varenicline has not been associated with rates of serum enzyme elevations during therapy greater than occurs with placebo therapy, but information on these abnormalities is limited and occasional instances of asymptomatic ALT elevations leading to drug discontinuation have been reported. In prelicensure pivotal registration trials in several thousand patients, varenicline was not associated with cases of jaundice or hepatitis. Since licensure, rare case reports of serum enzyme elevations without jaundice arising within 4 weeks of starting varenicline have been published, but largely in patients with other causes of liver injury (alcoholic liver disease, hepatitis C). The injury was self-limited in course and not associated with immunoallergic or autoimmune features. In Iceland, a single case of varenicline hepatotoxicity has been reported (Case 1), there having been an estimated 20,000 persons treated with the drug in the country since its introduction. Likelihood score: C (probable rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Varenicline is a partial nicotine agonist used orally to assist smoking cessation and by nasal spray for dry eyes. One researcher points out that based on animal data on nicotine, varenicline might interfere with normal infant lung development and recommends against its use in nursing mothers. Because no information is available on the use of varenicline during breastfeeding, an alternate drug is preferred, especially while nursing a newborn or preterm infant. However, maternal drug exposure after the nasal spray is only about 7.5% that of the oral drug, so the spray is much less likely to affect the infant. If a mother chooses to breastfeed while taking varenicline, she should monitor her infant for seizures and excessive vomiting. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Less than 20%. In vitro toxicity: CC₅₀ > 20 μM in RAW 264.7 macrophages, HUVECs, and human adrenal chromaffin cells [1][3][4] - In vivo acute toxicity: No mortality or obvious behavioral abnormalities (lethargy, ataxia) in mice treated with Varenicline dihydrochloride at oral doses up to 50 mg/kg [5] - Plasma protein binding rate: 10–20% (human plasma, ultrafiltration method) [2] |
| References |
[1]. Elif Baris, et al. Varenicline Prevents LPS-Induced Inflammatory Response via Nicotinic Acetylcholine Receptors in RAW 264.7 Macrophages. Front Mol Biosci. 2021 Oct 12;8:721533. [2]. Mihalak KB, et al. Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors.Mol Pharmacol. 2006 Sep;70(3):801-5. Epub 2006 Jun 9. [3]. Jin H, et al. Therapeutic concentrations of varenicline in the presence of nicotine increase action potential firing in human adrenal chromaffin cells. J Neurochem. 2017 Jan;140(1):37-52. [4]. Mitsuhisa Koga, et al. Varenicline promotes endothelial cell migration by lowering vascular endothelial-cadherin levels via the activated α7 nicotinic acetylcholine receptor-mitogen activated protein kinase axis. Toxicology. 2017 Sep 1;390:1-9. [5]. Bagdas D, et al. New insights on the effects of varenicline on nicotine reward, withdrawal and hyperalgesia in mice.Neuropharmacology. 2018 Aug;138:72-79. |
| Additional Infomation |
Varenicline is a prescription medication used to treat smoking addiction. This medication is the first approved nicotinic receptor partial agonist. Specifically, varenicline is a partial agonist of the alpha4/beta2 subtype of the nicotinic acetylcholine receptor. In addition it acts on alpha3/beta4 and weakly on alpha3beta2 and alpha6-containing receptors. A full agonism was displayed on alpha7-receptors. On March 9, 2015, the U.S. Food and Drug Administration warned that Varenicline, in the form of Pfizer Inc's quit-smoking drug, Chantix, has been associated with seizures and that some patients who drink while taking the drug may become aggressive or black out. Pfizer is conducting an additional safety study of the drug, results of which are expected in late 2015. The FDA said it is keeping the black box in place at least until the results of the trial are announced. Varenicline is a partial agonist of the nicotinic acetylcholine receptor and is used to help in smoking cessation. Varenicline has been associated with a low rate of serum enzyme elevations during therapy and, since approval and its widescale use, with rare instances of clinically apparent mild liver injury. Varenicline is a partial agonist of the nicotinic acetylcholine receptor (nAChR) subtype alpha4beta2. Nicotine stimulation of central alpha4beta2 nAChRs located at presynaptic terminals in the nucleus accumbens causes the release of the neurotransmitter dopamine, which may be associated with the experience of pleasure; nicotine addiction constitutes a physiologic dependence related to this dopaminergic reward system. As an AChR partial agonist, varenicline attenuates the craving and withdrawal symptoms that occur with abstinence from nicotine but is not habit-forming itself. A benzazepine derivative that functions as an ALPHA4-BETA2 NICOTINIC RECEPTOR partial agonist. It is used for SMOKING CESSATION. See also: Varenicline dihydrochloride (annotation moved to). Drug Indication For use as an aid in smoking cessation. Varenicline as a nasal spray is indicated for the symptomatic treatment of dry eye disease. FDA Label Mechanism of Action Varenicline is an alpha-4 beta-2 neuronal nicotinic acetylcholine receptor partial agonist. The drug shows high selectivity for this receptor subclass, relative to other nicotinic receptors (>500-fold alpha-3 beta-4, >3500-fold alpha-7, >20,000-fold alpha-1 beta gamma delta) or non-nicotinic receptors and transporters (>2000-fold). The drug competitively inhibits the ability of nicotine to bind to and activate the alpha-4 beta-2 receptor. The drug exerts mild agonistic activity at this site, though at a level much lower than nicotine; it is presumed that this activation eases withdrawal symptoms. Varenicline is a selective alpha4beta2 nicotinic acetylcholine receptor partial agonist. The drug binds with high affinity and selectivity to alpha4beta2 nicotinic acetylcholine receptors located in the brain and stimulates receptor-mediated activity, but at a substantially lower level than nicotine;1 6 this low-level receptor stimulation and subsequent moderate, sustained release of mesolimbic dopamine are thought to reduce craving and withdrawal symptoms associated with smoking cessation. Varenicline also blocks the ability of nicotine to activate alpha4beta2 receptors, preventing nicotine-induced stimulation of the mesolimbic dopaminergic system and thereby reducing the reinforcement and reward effects of cigarette smoking. ... The rationale for and the design of alpha(4)beta(2) neuronal nicotinic acetylcholine receptor (nAChR) partial agonists as novel treatments for tobacco addiction. Such agents are expected to exhibit a dual action by sufficiently stimulating alpha(4)beta(2)-nAChR-mediated dopamine release to reduce craving when quitting and by inhibiting nicotine reinforcement when smoking. Potent and selective alpha(4)beta(2) nAChR partial agonists that exhibit dual agonist and antagonist activity in preclinical models can be identified. The validity of this approach is demonstrated by the clinical efficacy of the alpha(4)beta(2) nAChR partial agonist varenicline, which has significantly better quit rates than do other treatments and offers a new option for smoking cessation pharmacotherapy. ... Varenicline has been shown to be a partial agonist of alpha4beta2 receptors, and in equilibrium binding assays, it is highly selective for the alpha4beta2 receptor. ... The functional activity of varenicline at a variety of rat neuronal nicotinic receptors expressed in Xenopus laevis oocytes and assayed under two-electrode voltage clamp /was examined/ . Varenicline is a potent, partial agonist at alpha4beta2 receptors, with an EC50 of 2.3 +/- 0.3 microM and an efficacy (relative to acetylcholine) of 13.4 +/- 0.4%. Varenicline has lower potency and higher efficacy at alpha3beta4 receptors, with an EC50 of 55 +/- 8 microM and an efficacy of 75 +/- 6%. Varenicline also seems to be a weak partial agonist at alpha3beta2 and alpha6-containing receptors, with an efficacy <10%. It is remarkable that varenicline is a potent, full agonist at alpha7 receptors with an EC50 of 18 +/- 6 microM and an efficacy of 93 +/- 7% (relative to acetylcholine). Thus, whereas varenicline is a partial agonist at some heteromeric neuronal nicotinic receptors, it is a full agonist at the homomeric alpha7 receptor. Some combination of these actions may be involved in the mechanism of varenicline as a smoking cessation aid. Varenicline dihydrochloride is a synthetic neuronal nicotinic acetylcholine receptor modulator, clinically used for smoking cessation [2][5] - Its anti-inflammatory mechanism in macrophages involves α7 nAChR activation, inhibiting NF-κB and MAPK signaling pathways to reduce pro-inflammatory cytokine production [1] - The compound promotes endothelial cell migration via α7 nAChR-mediated downregulation of VE-cadherin and activation of MAPK (ERK1/2, p38) [4] - In smoking cessation, it acts as a partial agonist at α4β2 nAChR (reducing nicotine craving) and full agonist at α7 nAChR (modulating reward pathways), while blocking nicotine binding to α4β2 [2][5] - It enhances action potential firing in adrenal chromaffin cells in the presence of nicotine, potentially modulating catecholamine release [3] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~62.5 mg/mL (~219.93 mM) |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.5189 mL | 17.5945 mL | 35.1890 mL | |
| 5 mM | 0.7038 mL | 3.5189 mL | 7.0378 mL | |
| 10 mM | 0.3519 mL | 1.7594 mL | 3.5189 mL |