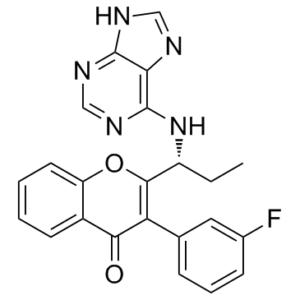
Tenalisib R Enantiomer is the R-isomer of Tenalisib (also known as RP-6530). Tenalisib is a new, potent and selective dual PI3Kδ/γ inhibitor with anticancer activity (IC50 = 24.5 nM and 33.2 nM for PI3Kδ and PI3Kγ, respectively). With selectivity over the α and β isoforms of more than 300-fold and 100-fold, respectively, it exhibits selective for PI3Kδ/γ It exhibits PI3K/ selectivity, with selectivity over the isoforms and being more than 300-fold and 100-fold, respectively. In both HEL-RS and HEL-RR cells, tenalisib shows a modest inhibition of proliferation (33-46% inhibition at 10 μM). Inhibition of proliferation is nearly 100% (>90% for HEL-RS and >70% for HEL-RR) when 10 μM tenalisib is added to ruxolitinib. The EC50 of ruxolitinib (5.8 μM ) in HEL-RR cells is significantly decreased by the addition of 5 μM tenalisib 4 hours before the addition of ruxolitinib.
Physicochemical Properties
| Molecular Formula | C23H18FN5O2 | |
| Molecular Weight | 415.42 | |
| Exact Mass | 415.144 | |
| Elemental Analysis | C, 66.50; H, 4.37; F, 4.57; N, 16.86; O, 7.70 | |
| CAS # | 1639417-54-1 | |
| Related CAS # | Tenalisib;1639417-53-0 | |
| PubChem CID | 117729511 | |
| Appearance | White to off-white solid | |
| LogP | 5.11 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 7 | |
| Rotatable Bond Count | 5 | |
| Heavy Atom Count | 31 | |
| Complexity | 704 | |
| Defined Atom Stereocenter Count | 1 | |
| SMILES | CC[C@H](C1=C(C(=O)C2=CC=CC=C2O1)C3=CC(=CC=C3)F)NC4=NC=NC5=C4NC=N5 |
|
| InChi Key | HDXDQPRPFRKGKZ-MRXNPFEDSA-N | |
| InChi Code | InChI=1S/C23H18FN5O2/c1-2-16(29-23-19-22(26-11-25-19)27-12-28-23)21-18(13-6-5-7-14(24)10-13)20(30)15-8-3-4-9-17(15)31-21/h3-12,16H,2H2,1H3,(H2,25,26,27,28,29)/t16-/m1/s1 | |
| Chemical Name | 3-(3-fluorophenyl)-2-[(1R)-1-(7H-purin-6-ylamino)propyl]chromen-4-one | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | PI3Kδ; PI3Kγ | ||
| ln Vitro |
Tenalisib is a dual PI3K δ/γ inhibitor, increases Ruxolitinib activity in JAK2-V617F mutant erythroleukemia cell lines[1]. Resistance to ruxolitinib was confirmed by a right-ward shift in EC50 of ruxolitinib in a HEL cell proliferation assay (0.82 μM Vs. 12.2 μM). Endogeous pAKT expression was 3.7-fold higher in HEL-RR compared to HEL-RS cells indicating activation of the AKT signaling pathway. While single-agent activity of RP6530 was modest (33-46% inhibition @ 10 μM) in both HEL-RS and HEL-RR cells, addition of 10 μM RP6530 to ruxolitinib was synergistic resulting in a near-complete inhibition of proliferation (>90% for HEL-RS and >70% for HEL-RR). While the order of addition did not affect the potency of RP6530, addition of 5 μM RP6530, 4 h prior to the addition of ruxolitinib resulted in a significant reduction in EC50 of ruxolitinib (5.8 μM) in HEL-RR cells. On lines with cell proliferation data, incubation of 10 μM RP6530 with ruxolitinib for 72 h increased the percent of apoptotic cells (55% in HEL-RS and 37% in HEL-RR) compared to either agent alone (16-27% in HEL-RS and 17-21% in HEL-RR). Conclusions: Ruxolitinib resistance in the V617F JAK-2 mutant HEL cells is accompanied by an increase in pAKT expression. Inhibition of pAKT via the addition of RP6530, a dual PI3K δ/γ inhibitor, resulted in a reversal of ruxolitinib resistance. Complementary activity was also observed in HEL-RS cells indicating that a combination of ruxolitinib and RP6530 could have a positive bearing on the clinical outcome in MF patients[1]. |
||
| ln Vivo |
|
||
| Cell Assay | Passive resistance was conferred by incubating HEL cells with increasing concentrations of ruxolitinib over an 8-10-week period. Endogenous JAK2, PI3Kδ, PI3Kδ, and pAKT were estimated by Western Blotting. RP6530, ruxolitinib, and the combination of RP6530 + Ruxolitinib were tested for their effect on viability and apoptosis. Cell viability was assessed by a MTT assay. Induction of apoptosis was analyzed by Annexin V/PI staining[1]. | ||
| Animal Protocol |
|
||
| References |
[1]. Abstract 2704: RP6530, a dual PI3K δ/γ inhibitor, potentiates ruxolitinib activity in the JAK2-V617F mutant erythroleukemia cell lines. |
||
| Additional Infomation | Background: Myelofibrosis (MF) represents a life-threatening neoplasm that manifests particularly in the elderly population and is characterized by bone marrow fibrosis and extramedullary hematopoeisis. While ruxolitinib, a JAK1/2 inhibitor, has recently been approved by the USFDA for its disease modifying potential in MF patients, it is still not considered as a curative option. Targeting another kinase such as PI3K, downstream of JAK, could therefore be a more efficient way of treating myelofibrotic neoplasms. RP6530 is a novel, potent, and selective PI3K δ/γ inhibitor that demonstrated high potency against PI3Kδ (IC50 = 25 nM) and γ (IC50 = 33 nM) enzymes with selectivity over α (>300-fold) and β (>100-fold) isoforms. The objective of this study was to evaluate the effect of a combination of ruxolitinib and RP6530 in the JAK2-V617F mutant Human Erythroleukemia (HEL) cell line. Methods: Passive resistance was conferred by incubating HEL cells with increasing concentrations of ruxolitinib over an 8-10-week period. Endogenous JAK2, PI3Kδ, PI3Kδ, and pAKT were estimated by Western Blotting. RP6530, ruxolitinib, and the combination of RP6530 + Ruxolitinib were tested for their effect on viability and apoptosis. Cell viability was assessed by a MTT assay. Induction of apoptosis was analyzed by Annexin V/PI staining. Results: Resistance to ruxolitinib was confirmed by a right-ward shift in EC50 of ruxolitinib in a HEL cell proliferation assay (0.82 μM Vs. 12.2 μM). Endogeous pAKT expression was 3.7-fold higher in HEL-RR compared to HEL-RS cells indicating activation of the AKT signaling pathway. While single-agent activity of RP6530 was modest (33-46% inhibition @ 10 μM) in both HEL-RS and HEL-RR cells, addition of 10 μM RP6530 to ruxolitinib was synergistic resulting in a near-complete inhibition of proliferation (>90% for HEL-RS and >70% for HEL-RR). While the order of addition did not affect the potency of RP6530, addition of 5 μM RP6530, 4 h prior to the addition of ruxolitinib resulted in a significant reduction in EC50 of ruxolitinib (5.8 μM) in HEL-RR cells. On lines with cell proliferation data, incubation of 10 μM RP6530 with ruxolitinib for 72 h increased the percent of apoptotic cells (55% in HEL-RS and 37% in HEL-RR) compared to either agent alone (16-27% in HEL-RS and 17-21% in HEL-RR). Conclusions: Ruxolitinib resistance in the V617F JAK-2 mutant HEL cells is accompanied by an increase in pAKT expression. Inhibition of pAKT via the addition of RP6530, a dual PI3K δ/γ inhibitor, resulted in a reversal of ruxolitinib resistance. Complementary activity was also observed in HEL-RS cells indicating that a combination of ruxolitinib and RP6530 could have a positive bearing on the clinical outcome in MF patients.[1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4072 mL | 12.0360 mL | 24.0720 mL | |
| 5 mM | 0.4814 mL | 2.4072 mL | 4.8144 mL | |
| 10 mM | 0.2407 mL | 1.2036 mL | 2.4072 mL |