Tegoprazan, a novel and potent P-CAB (potassium-competitive acid blocker), is a next-generation therapeutics developed for the treatment of acid-related gastrointestinal diseases such as gastroesophageal reflux disease (GERD) and peptic ulcers, acting by controling gastric acid secretion and motility.
Physicochemical Properties
| Molecular Formula | C20H19F2N3O3 |
| Molecular Weight | 387.37997174263 |
| Exact Mass | 387.139 |
| Elemental Analysis | C, 62.01; H, 4.94; F, 9.81; N, 10.85; O, 12.39 |
| CAS # | 942195-55-3 |
| Related CAS # | Tegoprazan-d6 |
| PubChem CID | 23582846 |
| Appearance | Off-white to light yellow solid powder |
| LogP | 3.2 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 3 |
| Heavy Atom Count | 28 |
| Complexity | 581 |
| Defined Atom Stereocenter Count | 1 |
| SMILES | O([C@H]1CCOC2=CC(=CC(=C12)F)F)C1C=C(C(=O)N(C)C)C=C2N=C(NC=12)C |
| InChi Key | CLIQCDHNPDMGSL-HNNXBMFYSA-N |
| InChi Code | InChI=1S/C20H19F2N3O3/c1-10-23-14-6-11(20(26)25(2)3)7-17(19(14)24-10)28-15-4-5-27-16-9-12(21)8-13(22)18(15)16/h6-9,15H,4-5H2,1-3H3,(H,23,24)/t15-/m0/s1 |
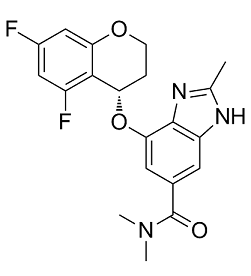
| Chemical Name | 7-[[(4S)-5,7-difluoro-3,4-dihydro-2H-chromen-4-yl]oxy]-N,N,2-trimethyl-3H-benzimidazole-5-carboxamide |
| Synonyms | Tegoprazan; 942195-55-3; Tegoprazan [INN]; CJ-12420; UNII-W017G7IF4S; W017G7IF4S; CJ 12420; CJ12420; RQ-00000004; RQ00000004; RQ 00000004;K-CAB; LXI-15028; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
H+/K+-ATPase (IC50 = 0.29~0.52 μM); H+/K+-ATPase (Ki = 0.52 μM for inhibition of porcine gastric H+/K+-ATPase) [3] |
| ln Vitro |
H+/K+-ATPase activity in pigs, dogs, and humans is inhibited by tigeprazan. Tegoprazan inhibits pH+/K+-ATPase in a reversible and potassium-competitive way. At tegoprazan doses up to 0.15 μM, 86% of H+/K+-ATPase activity is inhibited by 3 μM of tegoprazan [1]. Tegoprazan [(S)-4-((5,7-difluorochroman-4-yl)oxy)-N,N,2-trimethyl-1H-benzo[d]imidazole-6-carboxamide], a potassium-competitive acid blocker (P-CAB), is a novel potent and highly selective inhibitor of gastric H+/K+-ATPase. Tegoprazan inhibited porcine, canine, and human H+/K+-ATPases in vitro with IC50 values ranging from 0.29 to 0.52 μM, while that for canine kidney Na+/K+-ATPase was more than 100 μM. A kinetic analysis revealed that tegoprazan inhibited H+/K+-ATPase in a potassium-competitive manner and the binding was reversible [3]. - Tegoprazan inhibits porcine gastric H+/K+-ATPase activity in a concentration-dependent manner with a Ki of 0.52 μM, showing competitive inhibition with potassium ions. It also inhibits acid secretion in isolated rabbit gastric glands, reducing acid output with an EC50 of 0.37 μM [3] - In Caco-2 cell monolayers, Tegoprazan (10 μM) enhances gut barrier function by increasing transepithelial electrical resistance (TEER) and reducing paracellular permeability to FITC-dextran. It upregulates the expression of tight junction proteins (occludin, claudin-1, zonula occludens-1) at both mRNA and protein levels, as detected by qPCR and Western blot [1] |
| ln Vivo |
In dogs, egoprazan (1.0 mg/kg, po) effectively prevents stomach acidity brought on by histamine. The neutral range of pH is regained in the stomach after pentagastrin-induced acidification, with a dose of 1.0–3.0 mg/kg, po. In dogs administered with pentagastrin, egoprazan (3 mg/kg, face) causes immediate gastric phase III contractions of the migrating motor complex [1]. Inflammatory bowel disease (IBD) is a chronic immune-mediated disorder characterized by prolonged inflammation of the gastrointestinal tract. IBD can result from gut barrier dysfunction, altered gut microbiota, and abnormal intestinal immunity induced by environmental factors in genetically susceptible individuals. Proton pump inhibitors (PPIs) such as rabeprazole are frequently employed for gastric acid inhibition. However, long-term PPI administration can alter the intestinal microbiome composition, possibly worsening IBD severity. The present study revealed that tegoprazan, a potassium-competitive acid blocker, significantly improved colitis in mice and enhanced the intestinal epithelial barrier function. Tegoprazan alleviated gut microbiota dysbiosis and enhanced the growth of Bacteroides vulgatus. In turn, B. vulgatus alleviated intestinal inflammation by inhibiting epithelial adhesion of pathogenic bacteria. Unlike rabeprazole, tegoprazan did not induce gut dysbiosis. Our findings provide novel insights into the potential role of tegoprazan as an intestinal protectant for IBD and as a therapeutic agent for gastric acid-related diseases. [1] Tegoprazan, a novel potassium-competitive acid blocker (P-CAB), is a next-generation therapeutics developed for the treatment of acid-related gastrointestinal diseases such as gastroesophageal reflux disease (GERD) and peptic ulcers. In the present study, the in vitro and in vivo pharmacological properties of tegoprazan were compared with those of esomeprazole, a representative proton pump inhibitor. In vitro enzyme assays were performed using ion-leaky vesicles containing gastric H+/K+-ATPases isolated from pigs. The in vivo efficacies of tegoprazan were evaluated in rat models of GERD and peptic ulcer. Tegoprazan inhibited the activity of porcine H+/K+-ATPase with an IC50 value of 0.53 μM in a reversible manner, whereas esomeprazole showed weak and irreversible inhibition with an IC50 value of 42.52 μM. In a GERD model, tegoprazan showed dose-dependent efficacy in inhibiting esophageal injury and gastric acid secretion with an ED50 of 2.0 mg/kg, which was 15-fold more potent than that of esomeprazole. In peptic ulcer models, tegoprazan exhibited superior antiulcer activity compared with esomeprazole. The ED50 of tegoprazan in the naproxen-, ethanol-, and water-immersion restraint stress-induced peptic ulcer models were 0.1, 1.4, and 0.1 mg/kg, respectively. In the acetic acid-induced peptic ulcer model, the curative ratio of tegoprazan at 10 mg/kg was higher than that of esomeprazole at 30 mg/kg (44.2% vs. 32.7%, respectively), after 5 days of repeated oral administration. Thus, tegoprazan is a novel P-CAB that shows potent and reversible inhibition of gastric H+/K+-ATPase and may provide stronger efficacy compared with previous proton pump inhibitors.[2] Oral single administrations of tegoprazan ranging from 0.3 to 30 mg/kg in dogs were well absorbed into the blood stream and distributed in gastric tissue/fluid higher than in plasma. Tegoprazan potently inhibited histamine-induced gastric acid secretion in dogs, and a complete inhibition was observed at 1.0 mg/kg starting from 1 hour after administration. Moreover, an oral administration of tegoprazan at 1 and 3 mg/kg reversed the pentagastrin-induced acidified gastric pH to the neutral range. Interestingly, 3 mg/kg tegoprazan immediately evoked a gastric phase III contraction of the migrating motor complex in pentagastrin-treated dogs and similar effects was observed with the other P-CAB, vonoprazan. Tegoprazan is the novel P-CAB that may provide a new option for the therapy of gastric acid-related and motility-impaired diseases. [3] - In a rat model of acetic acid-induced colitis, oral administration of Tegoprazan (30 and 100 mg/kg/day) for 7 days reduces colonic damage, as indicated by lower disease activity index (DAI), decreased macroscopic score, and reduced myeloperoxidase (MPO) activity. It also improves gut barrier function by increasing colonic tight junction protein expression and reducing serum lipopolysaccharide (LPS) levels [1] - In pylorus-ligated rats, Tegoprazan (1, 3, 10 mg/kg, oral) dose-dependently inhibits basal gastric acid secretion, with a maximum inhibition of 94% at 10 mg/kg. It also reduces the volume and acidity of gastric juice [2] - In a rat model of water immersion stress-induced gastric lesions, Tegoprazan (3, 10, 30 mg/kg, oral) administered 1 hour before stress dose-dependently reduces lesion index, with 30 mg/kg showing 85% inhibition. Similar protective effects are observed in indomethacin-induced gastric lesions, with 10 mg/kg inhibiting lesion formation by 78% [2] - Tegoprazan (0.3-10 mg/kg, oral) reduces gastric motility in conscious rats, as measured by the transit of a radioactive marker. It also inhibits carbachol-induced gastric contractions in anesthetized rats in a dose-dependent manner [3] |
| Enzyme Assay |
Kinetic Analysis of H+/K+-ATPase Inhibition. [2] The enzyme kinetic study was performed based on the method of porcine ion-leaky assay described previously, except for the amount of vehicle (1 µg), and was tested with various concentrations of potassium (final concentrations of 2.0, 2.5, 3.5, 5.0, and 10 mM KCl) and tegoprazan (final concentrations of 0.15, 0.30, 0.45, and 0.60 µM). Measurement of Canine Kidney Na+/K+-ATPase Activity. [2] Canine kidney Na+/K+-ATPase enzyme was diluted to 7 mg/ml with 250 mM sucrose solution. The canine kidney Na+/K+-ATPase activity was measured in a 60 µl enzyme reaction mixture containing 11 µg of protein, test compound, 100 mM NaCl, 2 mM KCl, 3 mM MgSO4, 3 mM Na2ATP, and 40 mM Tris (pH 7.4 at 37°C) in a 96-well polystyrene plate. For 0% and 100% inhibition controls, the enzyme reaction was performed in the presence of 1% dimethylsulfoxide and 100 µM ouabain, respectively. The reaction was started by the addition of Na2ATP, the mixture was incubated for 30 minutes at 37°C, and then 30 µl of 10% SDS containing antifoam A was added to stop the reaction. The colorimetric analysis was performed in the same method for the porcine assay. Inhibition of Binding/Function against Pharmacologically Relevant Molecules. [2] Inhibitions of binding or functional activity against pharmacologically relevant molecules, receptors, ion channels, transporters, and enzymes, were tested in the presence of 10 µM tegoprazan. Regarding the Na+/K+-ATPase, tegoprazan was tested at 30 µM. All of the assays were conducted by Eurofins Cerep under their validated assay methods in the presence of standard inhibitors as an assay control (http://www.cerep.fr/cerep/users/pages/catalog/assay/catalog.asp). H+/K+-ATPase inhibition assay: Porcine gastric H+/K+-ATPase was isolated and incubated with various concentrations of Tegoprazan in the presence of ATP and potassium ions. ATP hydrolysis was measured by detecting inorganic phosphate release, and kinetic parameters (Ki) were calculated to assess competitive inhibition with potassium [3] |
| Cell Assay |
Cell Culture and Treatment [1] Human colon carcinoma cell lines, HT-29 and Caco-2 [HTB-37™, American Type Culture Collection (ATCC)], were maintained at 37°C in RPMI 1640 containing 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin-streptomycin solution in a humidified incubator with 5% CO2. Caco-2 cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% FBS and 1% penicillin-streptomycin solution. Cell viability was assessed by trypan blue staining under a microscope. Caco-2 cells were used to assess intestinal barrier function, with barrier damage induced by treatment with 40 ng/mL tumor necrosis factor (TNF)-α for 48 h. In Vitro Intestinal Permeability Assay [1] Next, to evaluate the intestinal epithelial barrier function, Caco-2 cells were plated into the upper chamber of the Transwell system (0.4 μm pore). Epithelial permeability was assessed by the analysis of transepithelial electrical resistance (TEER) and the paracellular flux of FITC-dextran (FD4). The electrical resistance of the Caco-2 cell monolayers cultured in the Transwell chamber was assessed using a Millicell-ERS instrument. FITC-dextran was added to the upper chamber at a final concentration of 1 mg/ml. Two hours after the addition of FITC-dextran, the medium from the lower chamber was collected, and fluorescence intensity was measured using a fluorescence microplate reader. - Gut barrier function assay in Caco-2 cells: Caco-2 cells were seeded on transwell inserts and differentiated into monolayers. Cells were treated with Tegoprazan (0.1-10 μM) for 24 hours. TEER was measured to assess barrier integrity, and paracellular permeability was evaluated using FITC-dextran flux. Tight junction protein expression was analyzed by qPCR (mRNA) and Western blot (protein) [1] - Isolated gastric gland assay: Rabbit gastric glands were isolated and incubated with Tegoprazan (0.01-10 μM) in the presence of stimulators (histamine, carbachol). Acid secretion was measured by the accumulation of a fluorescent dye sensitive to pH changes, and EC50 was determined [3] |
| Animal Protocol |
Measurement of Plasma and Gastric Juice Concentration of tegoprazan in Dogs. [3] Male beagle dogs at 8–11 months old were fasted overnight prior to dosing and dosed tegoprazan with 0.3, 3, and 30 mg/kg by mouth (n = 2) and blood samples were collected at 0.25, 0.5, 1, 2, 4, 8, and 24 hours post dosing. Male Heidenhain pouch (HP) dogs were orally dosed tegoprazan with 1 or 3 mg/kg, and blood and gastric juice were collected at 5 or 16 hours after the drug dose. Plasma and gastric concentrations of tegoprazan were quantified using liquid chromatography–tandem mass spectrometry (API4000 Triple Quadrupole mass spectrometer) and calculated using Analyst Program version 1.4.1. Pharmacokinetic parameters were determined with noncompartmental analysis using WinNonlin version 5.2.1 Measurement of Gastric Acid Secretion in the HP Dog Model. [3] A gastric pouch was constructed according to the method introduced by Heidenhain (1879). Briefly, male beagle dogs (7–12 kg body weight) were anesthetized with midazolam (0.2 mg/kg, i.m.) and medetomidine (0.05 mg/kg, i.m.) and isoflurane inhalation. The abdominal cavity was opened after the injection of atropine sulfate (0.5 mg/ml, 1 ml, i.m.) and infusion of Lactec D (100 ml/h). After exposing the stomach in the surgical field, a portion of the greater curvature opposite the splenic hilum was converted into a pouch with an adequate blood supply from the intact gastroepiploic artery. The main body of the stomach was reconstituted, while the pouch drained into an implanted metal cannula. After closing the pouch, the cannula was brought out of the abdominal cavity through the left lateral abdominal wall. Animals were allowed to recover from surgery for at least at last 3 weeks. The experiment was started after overnight fasting with free access to water. Acid secretion was stimulated by continuous intravenous infusion of histamine (80 μg/mg per hour) and maintained throughout the experimental periods. Gastric juice samples were collected by gravity drainage every 15 minutes throughout the experiment. At 90 minutes after initiation of the histamine infusion, tegoprazan, omeprazole, or vehicle (0.5% methylcellulose) was administrated orally. In the 5-day repeated dosing study, test drug or vehicle was administered orally for 5 days and its inhibitory effect on histamine-stimulated acid secretion was examined on treatment days 1 and 5. The collected gastric juice samples were centrifuged and acidity in the supernatant was determined using an automatic titrator. Gastric acid secretion at intervals of every 15 minutes (acidity × volume of the supernatant) was expressed as milliequivalents per 15 minutes. All results were calculated by MS EXCEL software In Vivo Intestinal Permeability Assay [1] Intestinal permeability was evaluated by measuring paracellular permeability to 4 kDa fluorescein isothiocyanate (FITC)-dextran on the day of sacrifice. Mice were orally administered 150 μL of 80 mg/mL FITC-dextran, and blood was collected 4 h after administration. Fluorescence intensity was measured using a fluorescent microplate reader - Acetic acid-induced colitis model in rats: Colitis was induced by intrarectal administration of acetic acid. Tegoprazan was suspended in 0.5% carboxymethyl cellulose (CMC) and administered orally at 30 or 100 mg/kg/day for 7 days, starting 1 day after induction. DAI was assessed daily, and on day 7, rats were sacrificed to collect colon tissues for macroscopic scoring, MPO activity measurement, and tight junction protein analysis [1] - Pylorus-ligated rat model: Rats were fasted for 24 hours, then pylorus-ligated under anesthesia. Tegoprazan (1, 3, 10 mg/kg) or vehicle (0.5% CMC) was administered orally immediately after ligation. Four hours later, gastric juice was collected to measure volume, acidity, and acid output [2] - Gastric lesion models: For water immersion stress-induced lesions, rats were fasted, administered Tegoprazan (3, 10, 30 mg/kg, oral) or vehicle, then subjected to water immersion stress for 7 hours. For indomethacin-induced lesions, rats were given Tegoprazan (3, 10 mg/kg) 30 minutes before indomethacin, and lesions were assessed 4 hours later. Lesion index was calculated based on the number and size of lesions [2] - Gastric motility studies: Conscious rats were fasted, administered Tegoprazan (0.3-10 mg/kg, oral), and 30 minutes later, a radioactive marker was given intragastrically. Gastric emptying and transit were measured by counting radioactivity in the stomach and intestines at 30 minutes. In anesthetized rats, gastric contractions were induced by carbachol, and Tegoprazan (0.1-1 mg/kg, intravenous) was administered to assess inhibition [3] |
| References |
[1]. Novel Potassium-Competitive Acid Blocker, Tegoprazan, Protects Against Colitis by Improving Gut Barrier Function. Front Immunol. 2022 May 25;13:870817. [2]. Effects of Tegoprazan, a Novel Potassium-Competitive Acid Blocker, on Rat Models of Gastric Acid-Related Disease. J Pharmacol Exp Ther. 2019 Jun;369(3):318-327. [3]. Tegoprazan, a Novel Potassium-Competitive Acid Blocker to Control Gastric Acid Secretion and Motility. J Pharmacol Exp Ther. 2018 Feb;364(2):275-286. |
| Additional Infomation |
Tegoprazan (also known as CJ-12420) is a novel therapeutic developed by CJ Healthcare Corp for treating acid-related gastrointestinal diseases. This drug is a potent and high-selective potassium-competitive acid blocker (P-CAB) with a fast onset of action and the ability to control gastric pH for a prolonged period of time. Tegoprazan’s strong and sustained effect is due to its ability to be slowly cleared from the gastric glands and exertion of effects independent of acid levels. It has also been observed to be efficacious independent of food intake. Mechanism of Action Tegoprazan works as a potassium-competitive acid blocker that is potent and highly selective. Its mechanism of action is different from that of the proton-pump inhibitors as this drug does not require conversion into an active form and can directly inhibit H+/K+‐ATPase in a reversible and K+‐competitive way. This is because it is an acid-resistant weak base with the ability to remain in the highly acidic canaliculi of gastric parietal cells. In conclusion, tegoprazan, a novel P-CAB, exhibits stronger and more reversible gastric acid suppression than traditional PPIs. However, the effect of tegoprazan on intestinal inflammation remains unknown. Long-term PPI administration reportedly increases and exacerbates the severity of IBD. To our knowledge, this study is the first to demonstrate that tegoprazan potentially ameliorates intestinal inflammation by enhancing intestinal epithelial barrier integrity, increasing the levels of Tregs, and modulating the composition of the gut microbiota, as demonstrated by in vitro and in vivo experiments. Additionally, we determined that tegoprazan promotes the growth of a particular member of commensal bacteria, B. vulgatus, which crucially contributes to the suppression of pathogenic microorganisms. Specifically, our findings provide critical insights into the potential treatment strategies using PPIs and P-CABs in IBD, although detailed mechanisms underlying the changes described warrant further characterization. [1] P-CAB is a new class of drugs for acid-related gastrointestinal diseases and is positioned as the next generation of PPIs. Clinical evaluation of vonoprazan has demonstrated its potent and long-acting efficacy in patients with gastroesophageal reflux disease and peptic ulcers compared with that of the currently available PPIs. In vitro and in vivo animal pharmacology studies of tegoprazan in this study have demonstrated that tegoprazan has similar pharmacological properties to those of vonoprazan. Tegoprazan is a novel P-CAB that may provide a new option for therapy in gastric acid-related diseases as well as in gastrointestinal motility-impaired diseases in clinical use.[3] Tegoprazan is a novel potassium-competitive acid blocker (P-CAB) that inhibits gastric H+/K+-ATPase by competing with potassium ions, thereby reducing gastric acid secretion. It shows rapid and potent effects in models of acid-related diseases and also exhibits protective effects in colitis by improving gut barrier function through upregulation of tight junction proteins [1][2][3] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~100 mg/mL (~258.14 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (6.45 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (6.45 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (6.45 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5814 mL | 12.9072 mL | 25.8144 mL | |
| 5 mM | 0.5163 mL | 2.5814 mL | 5.1629 mL | |
| 10 mM | 0.2581 mL | 1.2907 mL | 2.5814 mL |