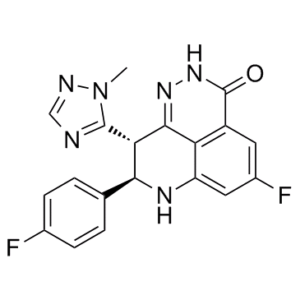
Talazoparib racemic (formerly known as BMN 673 and MDV 3800), the racemic mixture of talazoparib, is a potent PARP1/2 [poly(ADP-ribose) polymerase] inhibitor with favorable pharmacokinetic properties.
Physicochemical Properties
| Molecular Formula | C19H14F2N6O |
| Molecular Weight | 380.35 |
| Exact Mass | 380.12 |
| Elemental Analysis | C, 60.00; H, 3.71; F, 9.99; N, 22.10; O, 4.21 |
| CAS # | 1207456-00-5 |
| Related CAS # | 1207456-00-5; 1207456-01-6; 1207454-56-5 (racemic); 1373431-65-2 |
| PubChem CID | 135742498 |
| Appearance | White to off-white solid powder |
| LogP | 1.898 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 2 |
| Heavy Atom Count | 28 |
| Complexity | 654 |
| Defined Atom Stereocenter Count | 2 |
| SMILES | CN1C(=NC=N1)[C@H]2[C@@H](NC3=CC(=CC4=C3C2=NNC4=O)F)C5=CC=C(C=C5)F |
| InChi Key | HWGQMRYQVZSGDQ-HOTGVXAUSA-N |
| InChi Code | InChI=1S/C19H14F2N6O/c1-27-18(22-8-23-27)15-16(9-2-4-10(20)5-3-9)24-13-7-11(21)6-12-14(13)17(15)25-26-19(12)28/h2-8,15-16,24H,1H3,(H,26,28)/t15-,16-/m0/s1 |
| Chemical Name | (11R,12S)-7-fluoro-11-(4-fluorophenyl)-12-(2-methyl-1,2,4-triazol-3-yl)-2,3,10-triazatricyclo[7.3.1.05,13]trideca-1,5(13),6,8-tetraen-4-one |
| Synonyms | Talazoparib (8R,9S); (8R,9S)-LT-673; BMN 673 racemic; BMN673; BMN-673; LT673; LT 673; LT-673; MDV-3800; MDV 3800; MDV3800; trade name: Talzenna |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | PARP-1 ( IC50 = 144 nM ) | ||
| ln Vitro |
|
||
| ln Vivo |
|
||
| Enzyme Assay |
In order to determine the PARP inhibitor Ki, enzyme assays were carried out in 96-well FlashPlate using 0.5 U PARP1 enzyme, 0.25x activated DNA, 0.2 mCi [3H] NAD, and 5 mmol/L cold NAD (Sigma) in a final volume of 50 mL reaction buffer that contained 10% glycerol (v/v), 25 mmol/L HEPES, 12.5 mmol/L MgCl2, 50 mmol/L KCl, 1 mmol/L dithiothreitol (DTT), and 0.01% NP-40 (v/v), and pH 7.6. NAD was added to the PARP reaction mixture, either with or without inhibitors, to start the reaction, and it was then incubated for one minute at room temperature. The reaction was then stopped by adding 50 microliter of ice-cold 20% trichloroacetic acid (TCA) to each well. After the plate was sealed and shaken for an additional 120 minutes at room temperature, centrifugation was performed. Top-Count was used to determine the radioactive signal bound to the FlashPlate. The Michaelis-Menten equation was used to calculate PARP1 Km at different substrate concentrations (ranging from 1 to 100 mmol/L NAD). Using the formula Ki ¼ IC50/[1þ (substrate)/Km], compound Ki was computed from the enzyme inhibition curve. Using the same assay protocol, Km for the PARP2 enzyme and compound Ki were found. However, instead of using 30 ng of PARP2, 0.25x activated DNA, 0.2 mCi [3H] NAD, and 20 mmol/L cold NAD, the reaction was run for 30 minutes at room temperature. PARP enzyme assays[1] The ability of a test compound to inhibit PARP-1 enzyme activity was assessed using Trevigen’s PARP Assay Kit following manufacturer’s instruction. IC50 values were calculated using GraphPad Prism5 software. For PARP inhibitor Ki determination, enzyme assays were carried out in 96-well FlashPlate with 0.5 unit PARP1 enzyme, 0.25x activated DNA (Trevigen), 0.2 μCi [3H] NAD and 5 μM cold NAD in a final volume of 50 μL reaction buffer containing 10%glycerol(v/v), 25 mM Hepes, 12.5 mM MgCl2, 50 mM KCl, 1 mM DTT and 0.01% NP-40(v/v), pH 7.6. Reactions were initiated by adding NAD to the PARP reaction mixture with or without inhibitors and incubated for 1 min at room temperature. 50 μL of ice-cold 20% TCA was then added to each well to stop the reaction. The plate was sealed and shaken for a further 120 min at RT, followed by centrifugation. Radioactive signal bound to the FlashPlate was determined using TopCount. PARP1 Km was determined using Michaelis–Menten equation from various substrate concentrations (1-100 μM NAD). Compound Ki was calculated from enzyme inhibition curve according to the formula: Ki = IC50/(1+[substrate]/Km). Km for PARP2 enzyme and compound Ki were determined with the same assay protocol except 30 ng PARP2, 0.25x activated DNA, 0.2 μCi [3H] NAD and 20 μM cold NAD were used in the reaction for 30min at room temperature. Biacore binding assay[1] Recombinant human PARP1 (rhPARP1) catalytic domain (residues 662 – 1011) with N-terminal 6XHis-tag was generated in house and used in binding assay for PARP inhibitor interaction using Biacore T200 (GE Healthcare). rhPARP1 was immobilized on a CM5 sensor chip by amine coupling method. Briefly, one flow cell of a CM5 chip was first activated by a 7-min injection at 10 μL/min of freshly prepared 50 mM NHS: 200 mM EDC (1:1) at rate of 10 μL/min. Then rhPARP1 (100 μg/mL, in 10 mM MES pH 6.5) was injected onto the flow cell for 60-sec at 10 μL/min. The remaining active coupling sites were blocked with a 7-min injection of 1M ethanolamine at 10 μL/min. The immobilization buffer contains 10 mM Hepes pH 7.4, 150 mM NaCl, 0.05% Surfectant P20, 5 mM MgCl2, and 0.5 mM TCEP (tris(2-carboxyethyl)phosphine). The immobilization level was ~7600 RU. For binding kinetics measurement, PARP inhibitors at increasing concentrations (12.5, 25, 50, 100, 200 nM) were injected over the chip surface for 60 sec per injection. The exposure was followed by a dissociation phase of 3600 sec in running buffer (immobilization buffer + 1% DMSO) after the last injection. The flow rate was 50 μL/min. After sensorgrams were corrected for signals from a reference flow, kinetics was calculated with Biacore T200 evaluation software ver.1.0. Intracellular PAR formation assay[1] Cellular PAR synthesis assay assesses the ability of a test compound to inhibit polymerization of PAR. LoVo human colorectal tumor cells grown in 96-well microtiter plates overnight were pre-treated with increasing concentrations of PARP inhibitors for 30 min before H2O2 was added at a final concentration of 50 mM. After a 5-min treatment at room temperature, cells were fixed for 10 minutes with pre-chilled methanol/acetone(7:3) at −20 °C. Fixed cells were incubated with anti-PAR monoclonal antibody for 60 min, followed by incubation with FITC coupled goat anti-mouse IgG (diluted 1:100) and 1 μg/mL DAPI for 60 min. FITC signal was normalized with DAPI signal, and EC50 values were calculated using GraphPad Prism. |
||
| Cell Assay |
On a panel of eleven SCLC cell lines, BMN 673 has a strong inhibitory effect (IC50=1.7 to 15 nmol/L), all of which fall within ranges that are clinically feasible. Furthermore, there is a correlation between the expression of DNA repair proteins and the activity of the PI3K pathway and sensitivity to BMN673. Confocal microscopy[1] Cells were seeded on coverslips placed in 6-well plates and after 24 hours treated with several concentrations of olaparib or Talazoparib (BMN 673; MDV3800). 24 hours after treatment the cells were fixed in 10% formalin (3.7% PFA) for 1 hour. Cells were permeabilized with 0.2% Triton X-100 in PBS for 20 minutes, treated with 50 μL DNase I (diluted 1/10 in PBS) for 1 hour at 37°C and then blocked with IFF (PBS + 1% BSA and 2% FBS followed by filter sterilization) for 1 hour. The coverslips were then incubated with rabbit anti-γH2Ax primary (Millipore) and mouse anti-RAD51 primary (both 1:1000 in 50μL IFF) overnight at 4°C. The next day cells were incubated with anti-mouse Alexafluor 546 secondary and anti-rabbit Alexafluor 488 secondary (both 1:1000 in 50μL IFF) for one hour. Cells were then washed in PBS containing DAPI 1:10.000 for 10 minutes and attached on glass plates using Vectashield and nail polish. A minimum of four pictures were made of each coverslip using the Leica confocal microscope, and cells were subsequently counted. At least 100 cells were assessed per coverslip, being positive for γH2Ax if they had more than 5 foci per nucleus. The percentage of positive cells was plotted. |
||
| Animal Protocol |
|
||
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion After administration of talazoparib 1 mg orally once daily, the mean [% coefficient of variation (CV%)] AUC and maximum observed plasma concentration (Cmax) of talazoparib at steady-state was 208 (37%) ng x hr/mL and 16.4 (32%) ng/mL, respectively. The mean (CV%) steady-state Ctrough was 3.53 (61%) ng/mL. Steady state was reached within two to three weeks of therapy. The Tmax ranges from one to two hours. A high-fat, high-calorie food increased the mean Cmax by 46% and the median Tmax from one to four hours, without affecting the AUC. The major route of elimination is renal excretion. Approximately 68.7% of the total administered radiolabeled dose of talazoparib was recovered in urine, where 54.6% of that dose was in the form of an unchanged drug. About 19.7% of the drug was recovered in feces, with 13.6% of the dose is unchanged. The mean apparent volume of distribution of talazoparib is 420 L. The mean apparent oral clearance is 6.45 L/h. The inter-subject variability is 31%. Metabolism / Metabolites Talazoparib undergoes minimal hepatic metabolism. The metabolic pathways include mono-oxidation, dehydrogenation, cysteine conjugation of mono-desfluoro talazoparib, and glucuronide conjugation. Biological Half-Life The mean terminal plasma half-life (±standard deviation) is 90 (±58) hours in patients with cancer. |
||
| Toxicity/Toxicokinetics |
Hepatotoxicity Elevations in serum aminotransferase levels are common during talazoparib therapy occurring in 33% of patients, but rising above 5 times the upper limit of the normal range in only 1%. The elevations are generally transient and not associated with symptoms or jaundice. Furthermore, similar rates of aminotransferase elevations were reported in control, comparator arms. Talazoparib has had limited clinical use but has not been linked to instances of acute liver injury with symptoms or jaundice. Because of the limited clinical experience with using talazoparib and other PARP inhibitors, their potential for causing liver injury is not well defined. Likelihood score: E* (unproved but suspected cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the clinical use of talazoparib during breastfeeding. Because talazoparib is 74% bound to plasma proteins, the amount in milk is likely to be low. The manufacturer recommends that breastfeeding be discontinued during talazoparib therapy and for one month after the last dose. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding _In vitro_, the protein binding of talazoparib is 74% and is independent of talazoparib concentration. |
||
| References |
[1]. Clin Cancer Res . 2013 Sep 15;19(18):5003-15. [2]. Clin Cancer Res.2014Apr 15;20(8):2237. |
||
| Additional Infomation |
Talazoparib is an orally available small molecule inhibitor of the DNA repair enzyme poly ADP-ribose polymerase (PARP) which is used as an antineoplastic agent in the treatment of selected cases of breast cancer. Talazoparib is associated with a moderate rate of serum aminotransferase elevations during therapy and is suspected to cause rare instances of clinically apparent acute liver injury. Talazoparib is an inhibitor of mammalian polyadenosine 5’-diphosphoribose polymerases (PARPs), enzymes responsible for regulating essential cellular functions, such as DNA transcription and DNA repair. Developed by Pfizer, talazoparib was first approved by the FDA in October 2018 and by the EMA in June 2019. It was approved by Health Canada in September 2020. Talazoparib is currently used in the treatment of BRCA-mutated breast cancer and HRR-mutated prostate cancer. Talazoparib is a Poly(ADP-Ribose) Polymerase Inhibitor. The mechanism of action of talazoparib is as a Poly(ADP-Ribose) Polymerase Inhibitor. Talazoparib is an orally available small molecule inhibitor of the DNA repair enzyme poly ADP-ribose polymerase (PARP) which is used as an antineoplastic agent in the treatment of selected cases of breast cancer. Talazoparib is associated with a moderate rate of serum aminotransferase elevations during therapy and is suspected to cause rare instances of clinically apparent acute liver injury. Talazoparib is an orally bioavailable inhibitor of the nuclear enzyme poly(ADP-ribose) polymerase (PARP) with potential antineoplastic activity. Talazoparib selectively binds to PARP and prevents PARP-mediated DNA repair of single strand DNA breaks via the base-excision repair pathway. This enhances the accumulation of DNA strand breaks, promotes genomic instability and eventually leads to apoptosis. PARP catalyzes post-translational ADP-ribosylation of nuclear proteins that signal and recruit other proteins to repair damaged DNA and is activated by single-strand DNA breaks. See also: Talazoparib Tosylate (active moiety of). Drug Indication Talazoparib is indicated for the treatment of adult patients with deleterious or suspected deleterious germline BRCA-mutated (gBRCAm) HER2-negative locally advanced or metastatic breast cancer. This indication is approved by the FDA, EMA, and Health Canada. In the US, talazoparib is also indicated in combination with [enzalutamide] for the treatment of adult patients with HRR gene-mutated metastatic castration-resistant prostate cancer (mCRPC). Talzenna is indicated as monotherapy for the treatment of adult patients with germline BRCA1/2 mutations, who have HER2-negative locally advanced or metastatic breast cancer . Patients should have been previously treated with an anthracycline and/or a taxane in the (neo)adjuvant, locally advanced or metastatic setting unless patients were not suitable for these treatments. Patients with hormone receptor (HR)-positive breast cancer should have been treated with a prior endocrine-based therapy, or be considered unsuitable for endocrine-based therapy. Treatment of Ewing sarcoma Treatment of breast malignant neoplasms, Treatment of prostate malignant neoplasms Mechanism of Action Poly(ADP-ribose) polymerases (PARPs) are multifunctional enzymes involved in essential cellular functions, such as DNA transcription and DNA repair. PARPs recognize and repair DNA single-strand breaks (SSBs) via the base excision repair (BER) pathway. DNA double-strand breaks (DSBs) are repaired via homologous recombination by tumour suppressor proteins encoded by _BRCA1_ and _BRCA2_. Talazoparib is a potent inhibitor of poly (ADP-ribose) polymerase (PARP) enzymes, including PARP1 and PARP2. _In vitro_, talazoparib binds to PARP-1 and -2 isoforms with similar affinity. Inhibition of the BER pathway by talazoparib leads to the accumulation of unrepaired SSBs, which leads to the formation of DSBs, which is the most toxic form of DNA damage. While BRCA-dependent homologous recombination can repair DSBs in normal cells, this repair pathway is defective in cells with BRCA1/2 mutations, such as certain tumour cells. Inhibition of PARP in cancer cells with BRCA mutations leads to genomic instability and apoptotic cell death. This end result is also referred to as synthetic lethality, a phenomenon where the combination of two defects - inhibition of PARP activity and loss of DSB repair by HR - that are otherwise benign when alone leads to detrimental results. By inhibiting PARP, talazoparib increases the formation of PARP-DNA complexes resulting in DNA damage, decreased cell proliferation, and apoptosis. |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (6.57 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (6.57 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6292 mL | 13.1458 mL | 26.2916 mL | |
| 5 mM | 0.5258 mL | 2.6292 mL | 5.2583 mL | |
| 10 mM | 0.2629 mL | 1.3146 mL | 2.6292 mL |