Takeda-6d is a novel dual inhibitor of DFG-out RAF/vascular endothelial growth factor receptor 2 (VEGFR2) with anticancer activity. It inhibits wild-type B-RAF, mutant B-RAFV600E, and C-RAF (IC50s = 12, 7, and 1.5 nM, respectively), as well as VEGFR2 (IC50 = 2.8 nM). Takeda-6d (10 mg/kg) reduces tumor volume in an A375 melanoma mouse xenograft model.
Physicochemical Properties
| Molecular Formula | C27H19CLFN5O3S |
| Molecular Weight | 547.987866640091 |
| Exact Mass | 547.088 |
| CAS # | 1125632-93-0 |
| PubChem CID | 25206185 |
| Appearance | Typically exists as solid at room temperature |
| Density | 1.6±0.1 g/cm3 |
| Index of Refraction | 1.725 |
| LogP | 3.83 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 8 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 38 |
| Complexity | 974 |
| Defined Atom Stereocenter Count | 0 |
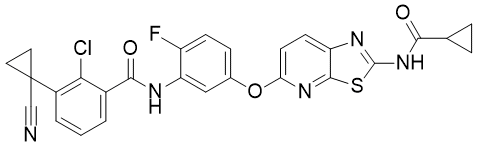
| SMILES | ClC1C(C(NC2C(=CC=C(C=2)OC2=CC=C3C(=N2)SC(=N3)NC(C2CC2)=O)F)=O)=CC=CC=1C1(C#N)CC1 |
| InChi Key | MDPMAXSABUPRJI-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C27H19ClFN5O3S/c28-22-16(2-1-3-17(22)27(13-30)10-11-27)24(36)31-20-12-15(6-7-18(20)29)37-21-9-8-19-25(33-21)38-26(32-19)34-23(35)14-4-5-14/h1-3,6-9,12,14H,4-5,10-11H2,(H,31,36)(H,32,34,35) |
| Chemical Name | 2-chloro-3-(1-cyanocyclopropyl)-N-[5-[[2-(cyclopropanecarbonylamino)-[1,3]thiazolo[5,4-b]pyridin-5-yl]oxy]-2-fluorophenyl]benzamide |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Takeda-6d targets BRAF(V600E) (IC50 = 0.9 nM), CRAF (IC50 = 3.2 nM), and VEGFR2 (IC50 = 1.8 nM) [1] |
| ln Vitro |
Against BRAF(V600E)-mutant A375 melanoma cells, Takeda-6d exhibited an antiproliferative effect with a GI50 value of 9.4 nM; in HT-29 colorectal cancer cells (wild-type BRAF), the GI50 was 37 nM [1] - Western blot analysis showed that Takeda-6d (10 nM, 24 h) significantly reduced the phosphorylation levels of ERK (p-ERK) in A375 cells and phosphorylation of AKT (p-AKT) in HUVECs, without affecting total ERK or AKT protein expression [1] - In HUVECs (human umbilical vein endothelial cells), Takeda-6d inhibited VEGF-induced cell proliferation with a GI50 of 8.7 nM and suppressed endothelial cell tube formation at concentrations ≥ 10 nM [1] |
| ln Vivo |
Rats with sufficient oral bioavailability (F = 70.5%) were given Takeda-6D (Compound 6d) (10 mg/kg, PO, once) [1]. In the rat A375 (BRAFV600E mutant) human melanoma xenograft model, Takeda-6D (0-10 mg/kg, PO, once) dramatically decreased ERK1 phosphorylation levels 24 hours after oral dose [1]. Takeda-6D (10 mg/kg, PO, twice daily for 2 weeks) shown no serious toxicity and tumor regression with a T/Cof of -7.0%; this should include both anti-angiogenic potency and BRAF inhibitory activity. Effectiveness [1]. In the A375 melanoma xenograft model in nude mice, oral administration of Takeda-6d at 30 mg/kg once daily for 21 days resulted in a tumor growth inhibition (TGI) rate of 78%; at 10 mg/kg, the TGI rate was 52% [1] - No significant reduction in body weight was observed in treated mice compared to the vehicle control group, indicating good in vivo tolerability at effective doses [1] |
| Enzyme Assay |
For BRAF(V600E), CRAF, and VEGFR2 kinase activity assays, a fluorescent-based peptide phosphorylation assay was used. The reaction mixture contained recombinant kinase, fluorescent-labeled substrate peptide, ATP (at Km concentration for each kinase), and serial dilutions of Takeda-6d. After incubation at 30°C for 60 minutes, the reaction was terminated by adding a stop solution containing a phosphorylation-specific antibody. Fluorescence intensity was measured, and IC50 values were calculated by fitting the dose-response curves [1] |
| Cell Assay |
Antiproliferative assay: Tumor cells (A375, HT-29) or HUVECs were seeded in 96-well plates at a density of 5×103 cells/well and incubated overnight. Serial dilutions of Takeda-6d were added, and cells were cultured for 72 hours. Cell viability was assessed using a colorimetric assay with a tetrazolium salt. GI50 values were determined as the compound concentration that inhibited cell growth by 50% compared to the vehicle control [1] - Western blot assay: A375 cells or HUVECs were seeded in 6-well plates and treated with Takeda-6d at specified concentrations for 24 hours. Cells were lysed in a buffer containing protease and phosphatase inhibitors. Protein extracts were separated by SDS-PAGE, transferred to PVDF membranes, and probed with primary antibodies against p-ERK, ERK, p-AKT, AKT, and β-actin (loading control). After incubation with horseradish peroxidase-conjugated secondary antibodies, chemiluminescent signals were detected and quantified [1] - Tube formation assay: HUVECs were seeded on Matrigel-coated 96-well plates and treated with Takeda-6d and VEGF (50 ng/mL). After 6 hours of incubation, tube formation was visualized under a microscope, and the number of tube branches was counted to evaluate anti-angiogenic activity [1] |
| Animal Protocol |
Female nude mice (6-8 weeks old) were subcutaneously inoculated with 5×106 A375 cells into the right flank. When tumors reached an average volume of 150 mm3, mice were randomly divided into three groups (n=6 per group): vehicle control, Takeda-6d 10 mg/kg, and Takeda-6d 30 mg/kg. Takeda-6d was formulated in 0.5% carboxymethylcellulose sodium (CMC-Na) aqueous solution and administered via oral gavage once daily for 21 consecutive days. Tumor volume (measured using calipers, volume = length × width2 / 2) and body weight were recorded every 3 days. At the end of the study, mice were euthanized, and tumors were excised and weighed [1] |
| ADME/Pharmacokinetics |
In mice, after intravenous administration of Takeda-6d at 5 mg/kg, the terminal half-life (t1/2) was 2.8 hours, and the area under the plasma concentration-time curve (AUC0-∞) was 12.6 μg·h/mL. After oral administration at 30 mg/kg, the AUC0-∞ was 28.9 μg·h/mL, corresponding to an oral bioavailability of 46% [1] - Plasma protein binding of Takeda-6d was determined to be 92% in mouse plasma and 94% in human plasma [1] |
| Toxicity/Toxicokinetics |
In the 21-day in vivo efficacy study, Takeda-6d at doses up to 30 mg/kg (oral) did not cause significant changes in mouse body weight (≤5% weight loss compared to control) or obvious histopathological abnormalities in major organs (liver, kidney, heart, lung, spleen) [1] - No significant inhibition of cytochrome P450 isoforms (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4) was observed with Takeda-6d at concentrations up to 10 μM in human liver microsomes [1] |
| References |
[1]. Design and synthesis of novel DFG-out RAF/vascular endothelial growth factor receptor 2 (VEGFR2) inhibitors. 1. Exploration of [5,6]-fused bicyclic scaffolds. J Med Chem. 2012 Apr 12;55(7):3452-78. |
| Additional Infomation |
Takeda-6d is a dual inhibitor of RAF kinases (BRAF(V600E), CRAF) and VEGFR2, binding to the DFG-out conformation of both targets [1] - Its dual mechanism of action simultaneously suppresses tumor cell proliferation (via RAF-MEK-ERK pathway inhibition) and tumor angiogenesis (via VEGFR2-AKT pathway inhibition), leading to synergistic antitumor effects [1] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.8249 mL | 9.1243 mL | 18.2485 mL | |
| 5 mM | 0.3650 mL | 1.8249 mL | 3.6497 mL | |
| 10 mM | 0.1825 mL | 0.9124 mL | 1.8249 mL |