TC-Mps1-12 is a novel, selective and potent monopolar spindle 1 (Mps1) inhibitor with anticancer activity. With an IC50 of 6.4 nM, it inhibits Mps1. In the A549 lung cancer xenograft model, it shows good cellular activity, selectivity, pharmacokinetic properties, and efficacy.
Physicochemical Properties
| Molecular Formula | C17H20N6O |
| Molecular Weight | 324.380302429199 |
| Exact Mass | 324.169 |
| Elemental Analysis | C, 62.95; H, 6.21; N, 25.91; O, 4.93 |
| CAS # | 1206170-62-8 |
| Related CAS # | 1206170-62-8 |
| PubChem CID | 70682875 |
| Appearance | White to off-white solid powder |
| LogP | 2.5 |
| Hydrogen Bond Donor Count | 4 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 24 |
| Complexity | 485 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | XDEFNAWAKYQBQY-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C17H20N6O/c1-17(2,3)23-16-12(9-18)13(19)8-14(22-16)21-11-6-4-10(5-7-11)15(20)24/h4-8H,1-3H3,(H2,20,24)(H4,19,21,22,23) |
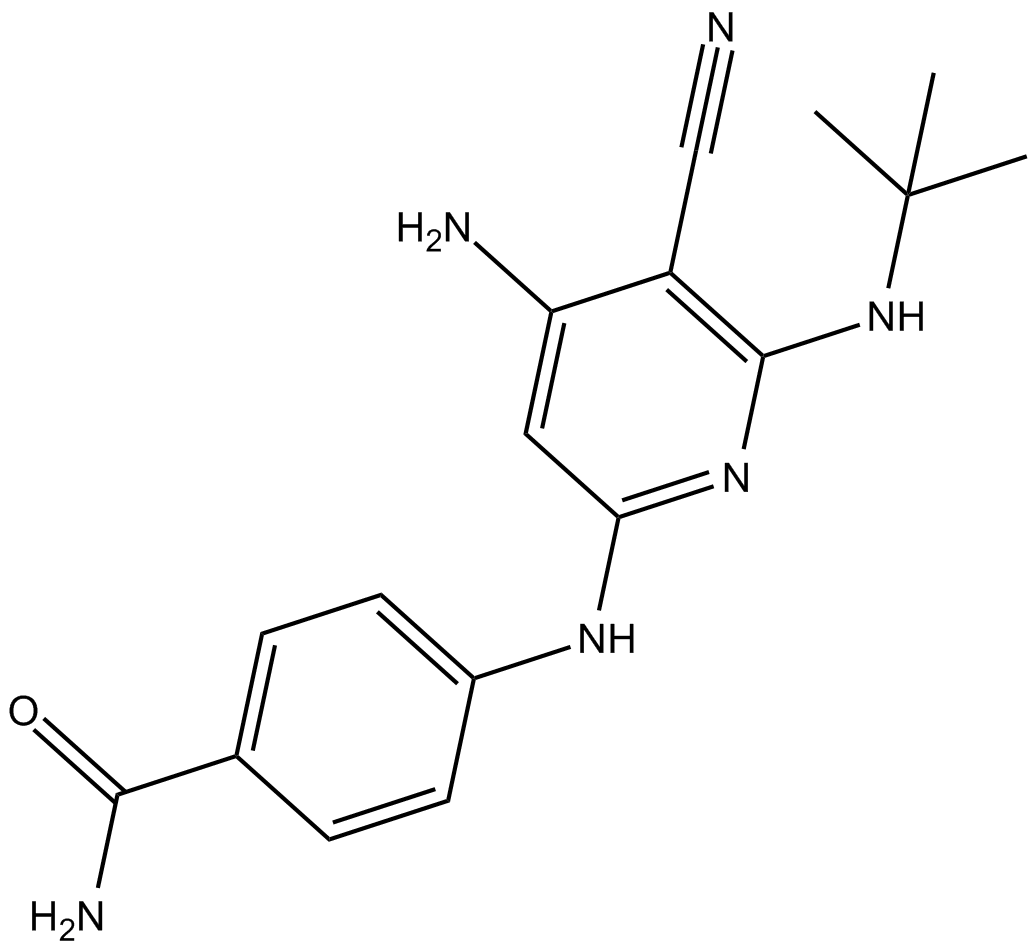
| Chemical Name | 4-[[4-amino-6-(tert-butylamino)-5-cyanopyridin-2-yl]amino]benzamide |
| Synonyms | TCMps112; TC Mps1 12; TC-Mps1-12 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | Mps1 (IC50 = 6.4 nM) |
| ln Vitro |
TC-Mps1-12 inhibits the growth of pMps1 cell lines in the autophosphorylation assay, with IC50 values of 131 nM[1]. TC-Mps1-12 (72 hours) suppresses cell growth with an IC50 value of 0.84 μM and in a dose-dependent manner[1]. Next, we turned our attention to the C2 side chain of the pyridine ring to further improve enzymatic activity and selectivity over JNK1. The crystal structure revealed that the C2 side chain of the ethyl ether lies in the ribose pocket that consists of hydrophobic residues Val539 and Ile663. Therefore, we examined a small set of hydrophobic substituents and found that substitution with a t-butylamino group (TC-Mps1-12) led to a 6-fold improvement in potency with a 36-fold selectivity for Mps1 over JNK1[1]. Once the highly potent Mps1 inhibitors 9 and 12/TC-Mps1-12 were identified, we examined the cellular effects of the compounds (Table 2). To investigate the cellular inhibition of Mps1, we developed an autophosphorylation assay using a cell line that stably expresses FLAG-tagged Mps1 under the control of a tetracycline (Tet)-suppressible promoter. TC-Mps1-12 showed an IC50 value of 131 nM, while 9 showed weak inhibition (1.8 μM) in this autophosphorylation assay. These antiproliferative activities were also examined in A549 lung carcinoma cell lines. Compounds 9 and TC-Mps1-12 inhibited the growth of these cell lines with IC50 values of 9.5 and 0.84 μM, respectively. The antiproliferative effects correlated with the cellular inhibition of Mps1[1]. To evaluate kinase selectivity, compound TC-Mps1-12 was screened against a panel of 95 kinases. We found that TC-Mps1-12 displayed an excellent selectivity profile in this panel, except for the Flt3 and Flt3 mutants (D835Y) (50 and 86% inhibition at 1 μM, respectively; Supporting Information). This selectivity profile can be explained by the crystal structure of the enzyme and the inhibitor complex. Our compounds form hydrogen bonds with the flipped carbonyl of Cys604 next to Gly605 and the carbonyl of Glu603 at the hinge region in an unusual manner. Therefore, our Mps1 inhibitors do not have the ability to bind to the “usual” hinge regions of other kinases, which accounts for the excellent kinase selectivity of TC-Mps1-12. |
| ln Vivo |
C-Mps1-12 (25-100 mg/kg; p.o.; daily; for 19 days) inhibits A549 cell growth in a dose-dependent way in vivo. TC-Mps1-12 inhibits tumor growth by 47% at a dose of 100 mg/kg without causing weight loss[1]. TC-Mps1-12 TC-Mps1-12 exhibits good PK properties, with a Cmax of 3542 ng/mL and an AUC of 6604 ng h/mL at an oral dose of 25 mg/kg[1]. |
| Enzyme Assay |
Mps1 enzyme assay. [1] Mps1 kinase activity was measured by the DELFIA® assay that monitors phosphorylation of the p38 MAPK peptide (biotin-AGAGLARHTDDEMTGYVA) using a phosphorylated site specific antibody. Test compounds were prepared as 10 mM solutions in DMSO and diluted with water as required to give a range of final assay conditions. Test compounds (1 µL, 10% DMSO), Mps1 solution (5 µL) (consisting of 0.5 µg/mL full length Mps1, 25 mM Tris-HCl (pH 7.5), 5 mM β-glycerophosphate, 0.1 mM Na3VO4, 5 mM MgCl2, and 2 mmol/L DTT) and substrate solution (5 µL) (consisting of 30 µM p38 MAPK peptide, 30 µM ATP, 25 mM Tris-HCl (pH 7.5), 5 mM βglycerophosphate, 2 mM DTT, 0.1 mM Na3VO4, 5 mM MgCl2, and 0.1%(w/v) BSA) were added to wells of a 384-well plate. The plate was incubated at 25°C for 90 min. Each reaction was stopped by the addition of a quenching buffer consisting of 25 mM of Tris HCl (pH 7.5), 100 mM of EDTA, 0.01%(v/v) of TritonX-100, and 0.1%(w/v) of BSA. An aliquot of 2.4 µL of the solution was transferred into another 384 well-plate to which the quenching buffer (60 µL) was added. An aliquot of 50 µL of the solution was transferred into a black NeutrAvidin-coated plate and incubated for 30 min at room temperature. The plate was washed three times with 100 µL of TTBS (10 mM Tris, 40 mM TrisHCl, 150 mM NaCl, and 0.05% (v/v) Tween20). This plate was added 50 µL of anti-p38 (phospho T180) antibody solution and incubated for 2 h at room temperature. The plate was washed five times with TTBS (100 µL) and 50 µL of europium-labeled anti-rabbit IgG was added and then incubated for 2 h at rt. The plate was washed five times with TTBS (100 µL), 50 µL of enhancement solution was added and then incubated for 5 min at rt. The plate was read on an ARVO multilabel counter using a time-resolved measurement mode reading fluorescence at 615 nm. The concentration of test compounds required to inhibit Mps1 activity by 50% was calculated as IC50. JNK1 enzyme assay. [1] Inhibition of JNK1 kinase activity by the test compounds was measured by phosphorylation of the JNK1 substrate ATF2 using the enzyme-linked immunosorbent assay (ELISA). Test compounds (10 mM in DMSO) diluted serially with DMSO were further diluted with working solution (consistent of TBS (10 mM Tris, 40 mM Tris-HCl, 150 mM NaCl, pH = 7.6), 0.1% (w/v) BSA, 10 mM MgCl, 5 mM MnCl, 0.2 mM Na3VO4 and 1 mM DTT) to prepare as test compounds solution (10% DMSO). ATP solution, biotinilated-ATF2 substrate solution and JNK1 enzyme solution diluted with working solution were prepared. The enzymatic reaction was performed in 384-well plates in a final volume of 55 µL. Five µL of test compound solution, 20 µL of substrate solution (final concentration: 50 nM) and 20 µL of enzyme solution (JNK1 kinase activity was linear with respect to each enzyme lot) were added to a 384-well plate and the enzymatic reaction was initiated by adding 10 µL of ATP solution (final concentration: 0.2 µM). After the plate has been incubated for 1 h at room temperature, the enzymatic reaction was stopped by adding 5 µL of 0.5 M EDTA solution (pH = 8.0). An aliquot of 40 µL was transferred into a white NeutrAvidincoated 384-well plate and incubated for 0.5 h at room temperature. The plate was washed three times with 100 µL of TTBS (10 mM Tris, 40 mM Tris-HCl, 150 mM NaCl, and 0.05% (v/v) Tween20). This plate was treated with 40 µL of anti-phospho-ATF2 (phospho Thr 69, 71) antibody 1:500 dilution with TBS containing 0.1% (w/v) BSA and incubated for 1.5 h at room temperature. The plate was washed three times with TTBS (100 µL) and 40 µL of HRP conjugated antimouse IgG 1:20000 dilution with TBS containing 0.1% (w/v) BSA was added and then incubated for 1.5 h at room temperature. The plate was washed three times with 100 µL of TTBS and the phosphorylated ATF2 was detected using colorimetric ELISA adding 40 µL of TMB solution for 5 min at room temperature. The colorimetric reaction was stopped by adding 40 µL of 0.5 M H2SO4 solution, and then 40 µL of solutions were transferred into a new clear-bottom 384-plate. The plate was read on ARVO multilabel counter using a photometric measurement mode reading absorbance at 450 nm. The concentration of test compounds required to inhibit JNK1 activity by 50% was calculated as IC50. |
| Cell Assay |
Cell Line: 549 cells Incubation Time: 72 hours Result: Inhibited the growth of cells in a dose-dependent manner and with an IC50 values of 0.84 μM. Cell proliferation assay. [1] This assay was conducted using the A549 human lung adenocarcinoma epithelial cell line provided by ATCC. A549 cells were cultured in DMEM media S9 supplemented with 10% fetal bovine serum, 50 units/mL penicillin and 50 µg/mL streptomycin at 37°C under 5% CO2. The A549 cells were seeded in a 96-well plate at 1000 cells/100 µL /well and incubated overnight. The test compound (10 mM in DMSO) was diluted 500 times with the culture medium followed by further nine-step dilution by twofold. The same amounts of the seriallydiluted compounds as the cell culture volume were transferred to the 96-well plate containing the cells. After incubation at 37°C under 5% CO2 for 72 h, cells were added to 10 µL of WST-8, and the optical densities at 450 nm and 650 nm were quantified using an Emax microplate reader. The IC50 value was determined as the concentration of compound required to reduce cell viability by 50% of the DMSO control. Mps1 autophosphorylation assay. [1] RERF-LC-AI Tet-off cells expressing FLAG Mps1 (in-house, RERF-LC-AI cells were provided by RIKEN) were maintained in DMEM media supplemented with 10% Tet System Approved FBS, 50 units/mL penicillin and 50 µg/mL streptmycin, 300 µg/mL G418 and 1 µg/mL doxycycline at 37°C under 5% CO2. The cells were seeded in 15 cm dish at density of 2.8 × 105 cells/dish in the culture media in the absence of G418 and doxycycline, and cultured for 5 h. After washing with PBS (-), the cells were cultured in the doxycycline-free medium for 3 days. The cells were reseeded in a 24-well plate at 2.8 × 104 cells/well and cultured overnight. The test compound (10 mM in DMSO) diluted serially in the doxycycline-free medium was used to treat the cells in the 24-well plate for 3 h. The cells were washed with PBS (-), then lysed with 1×SDS sample buffer (100 µL/well) followed by denaturation at 98°C for 10 min. The cell lysates were resolved on SDS-PAGE (6% acrylamide), transferred onto a PVDF membrane which, in turn, was blocked in 1% BlockAce and incubated with anti-FLAG M2 antibody. The membrane was treated with HRP-conjugated antimouse IgG. FLAG-tagged Mps1 was detected using ECL Plus and LAS-3000. The phosphorylated form was discriminated from the unphosphorylated form by the band shift. The density of each band was quantified using Multigauge. The inhibitory activity of the test compound was represented by the percentage of the band density in the DMSO- S10 treated sample. The concentration of the test compound required to inhibit the autophosphorylation of FLAG-Mps1 by 50% was calculated as IC50 |
| Animal Protocol |
A549 mouse xenograft model 25 mg/kg, 50 mg/kg, 100 mg/kg Oral administration; once daily; for 19 days In vivo antitumor assay.[1] A549 cells were cultured in the medium described above. A549 cells (1 × 107 cells) were inoculated subcutaneously into the flank of female nude mice (BALB/cA Jcl-nu/nu, 7 weeks old). The treatment was started when the tumor volumes reached around 200 mm3 . The test compound/TC-Mps1-12 suspended in 0.5% methylcellulose solution was administered p.o. daily for 19 days (n = 6). Tumors were measured with a caliper to calculate tumor volume formulas as follows: tumor volume = (length × width × width)/2. Tumor sizes and mouse body weights were recorded on days 2, 5, 9, 12, 16 and 19. Antitumor activity was expressed as T/C (%): (the mean tumor volume of treated animals/mean tumor volume of control animals) × 100. [1] |
| ADME/Pharmacokinetics | The pharmacokinetic (PK) profile of TC-Mps1-12 was studied using mice (Table 2). TC-Mps1-12/Compound 12 showed good PK properties with a Cmax of 3542 ng/mL and AUC of 6604 ng h/mL at an oral dose of 25 mg/kg. Encouraged by these results, the in vivo efficacy of 12 was examined with the A549 xenograft model. Mice bearing xenografts were dosed orally once daily with an escalating dose from 25 to 100 mg/kg for 19 days. The results showed that 12 inhibited the growth of A549 cells in a dose-dependent manner (Figure 5). At a dose of 100 mg/kg, 12 exhibited 47% tumor growth inhibition without body weight loss. [1] |
| References |
[1]. Diaminopyridine-based potent and selective mps1 kinase inhibitors binding to an unusual flipped-Peptide conformation. ACS Med Chem Lett. 2012 Jun 6;3(7):560-4. |
| Additional Infomation |
Monopolar spindle 1 (Mps1) is an attractive cancer drug target due to the important role that it plays in centrosome duplication, the spindle assembly checkpoint, and the maintenance of chromosomal stability. A design based on JNK inhibitors with an aminopyridine scaffold and subsequent modifications identified diaminopyridine 9 with an IC50 of 37 nM. The X-ray structure of 9 revealed that the Cys604 carbonyl group of the hinge region flips to form a hydrogen bond with the aniline NH group in 9. Further optimization of 9 led to 12 with improved cellular activity, suitable pharmacokinetic profiles, and good in vivo efficacy in the mouse A549 xenograft model. Moreover, 12 displayed excellent selectivity over 95 kinases, indicating the contribution of its unusual flipped-peptide conformation to its selectivity. [1] In conclusion, potent and selective diaminopyridine-based Mps1 inhibitors have been successfully identified. X-ray crystallographic data indicate that the Cys604 carbonyl group of the hinge region flips to form a hydrogen bond with the aniline NH group at the 6-position of pyridine. The excellent kinase selectivity of the optimized compound 12 can be explained by the unusual peptide flip at the hinge region. The diaminopyridine scaffold should be a good template for more advanced inhibitors of Mps1. Diaminopyridine 12 also exhibited good cellular activity and pharmacokinetic properties and was efficacious in the A549 lung cancer xenograft model. Additionally, 12 has good leadlike profiles: low MW (324), CLog P (2.8), and high LE (0.47). The selective Mps1 inhibitor 12 with in vivo efficacy could be used as a valuable tool for elucidating the biological functions in cancer therapy.[1] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.0828 mL | 15.4140 mL | 30.8280 mL | |
| 5 mM | 0.6166 mL | 3.0828 mL | 6.1656 mL | |
| 10 mM | 0.3083 mL | 1.5414 mL | 3.0828 mL |