SMCC-DM1 (DM1-SMCC) is DM1 (mertansine) with a reactive linker SMCC to synthesize antibody drug conjugate. DM1 (mertansine), a thiol-containing maytansinoid, is a potent microtubule-disrupting agent. DM1 is an antibody-conjugatable maytansinoid that was developed to overcome systemic toxicity associated with maytansine and to enhance tumor-specific delivery. DM1 binds at the tips of microtubules and suppresses the dynamicity of microtubules.
Physicochemical Properties
| Molecular Formula | C51H66CLN5O16S |
| Molecular Weight | 1072.6116528511 |
| Exact Mass | 1071.391 |
| Elemental Analysis | C, 57.11; H, 6.20; Cl, 3.30; N, 6.53; O, 23.87; S, 2.99 |
| CAS # | 1228105-51-8 |
| PubChem CID | 92131096 |
| Appearance | White to off-white solid powder |
| Density | 1.4±0.1 g/cm3 |
| Index of Refraction | 1.626 |
| LogP | 3.28 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 17 |
| Rotatable Bond Count | 15 |
| Heavy Atom Count | 74 |
| Complexity | 2250 |
| Defined Atom Stereocenter Count | 8 |
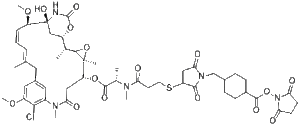
| SMILES | ClC1C(=CC2CC(C)=CC=CC([C@]3(C[C@@H]([C@@H](C)C4[C@](C)(C(CC(N(C)C=1C=2)=O)OC([C@H](C)N(C)C(CCSC1CC(N(C1=O)CC1CCC(C(=O)ON2C(CCC2=O)=O)CC1)=O)=O)=O)O4)OC(N3)=O)O)OC)OC |t:7,9| |
| InChi Key | IADUWZMNTKHTIN-IOBAKXROSA-N |
| InChi Code | InChI=1S/C51H66ClN5O16S/c1-27-10-9-11-37(69-8)51(67)25-35(70-49(66)53-51)28(2)45-50(4,72-45)38(24-42(61)55(6)33-21-31(20-27)22-34(68-7)44(33)52)71-47(64)29(3)54(5)39(58)18-19-74-36-23-43(62)56(46(36)63)26-30-12-14-32(15-13-30)48(65)73-57-40(59)16-17-41(57)60/h9-11,21-22,28-30,32,35-38,45,67H,12-20,23-26H2,1-8H3,(H,53,66)/b11-9-,27-10+/t28-,29+,30?,32?,35+,36?,37-,38-,45?,50+,51+/m1/s1 |
| Chemical Name | 2,5-dioxopyrrolidin-1-yl 4-((3-((3-(((2S)-1-(((14S,16S,33S,2R,4R,10E,12Z,14R)-86-chloro-14-hydroxy-85,14-dimethoxy-33,2,7,10-tetramethyl-12,6-dioxo-7-aza-1(6,4)-oxazinana-3(2,3)-oxirana-8(1,3)-benzenacyclotetradecaphane-10,12-dien-4-yl)oxy)-1-oxopropan-2-yl)(methyl)amino)-3-oxopropyl)thio)-2,5-dioxopyrrolidin-1-yl)methyl)cyclohexane-1-carboxylate |
| Synonyms | DM1-SMCC; DM1 SMCC; DM1SMCC; DM1-SMCC; SCHEMBL20153009; IADUWZMNTKHTIN-MLSWMBHTSA-N; 1613362-80-3; SMCC-DM1 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment (e.g. under nitrogen), avoid exposure to moisture. |
| Shipping Condition | Ship with dry ice. |
Biological Activity
| Targets | Maytansinoids; Tubulin |
| ln Vitro |
HCC1954 and MDA-MB-468 cell proliferation is inhibited by SMCC-DM1, with IC50 values of 17.2 and 49.9 nM, respectively[1]. Antibody-drug conjugates are targeted anticancer agents consisting of a cytotoxic drug covalently linked to a monoclonal antibody for tumor antigen–specific activity. Once bound to the target cell-surface antigen, the conjugate must be processed to release an active form of the drug, which can reach its intracellular target. Here, researchers used both biological and biochemical methods to better define this process for antibody-maytansinoid conjugates. In particular, researchers examined the metabolic fate in cells of huC242-maytansinoid conjugates containing either a disulfide linker (huC242-SPDB-DM4) or a thioether linker (huC242-SMCC-DM1). Using cell cycle analysis combined with lysosomal inhibitors, we showed that lysosomal processing is required for the activity of antibody-maytansinoid conjugates, irrespective of the linker. We also identified and characterized the released maytansinoid molecules from these conjugates, and measured their rate of release compared with the kinetics of cell cycle arrest. Both conjugates are efficiently degraded in lysosomes to yield metabolites consisting of the intact maytansinoid drug and linker attached to lysine. The lysine adduct is the sole metabolite from the thioether-linked conjugate. However, the lysine metabolite generated from the disulfide-linked conjugate is reduced and S-methylated to yield the lipophilic and potently cytotoxic metabolite, S-methyl-DM4. These findings provide insight into the mechanism of action of antibody-maytansinoid conjugates in general, and more specifically, identify a biochemical mechanism that may account for the significantly enhanced antitumor efficacy observed with disulfide-linked conjugates.[3] |
| ln Vivo | The in vivo efficacy of nBT062-SPDB-DM4, nBT062-SMCC-DM1, and nBT062-SPP-DM1 was next evaluated in SCID mice bearing established CD138-positive MOLP-8 human MM cells. A single i.v. administration of the immunoconjugates caused significant dose-dependent tumor growth inhibition and tumor regression at concentrations that were well tolerated, evidenced by stable body weight. nBT062-SPDB-DM4 was the most active conjugate tested in this model. In addition, weekly dosing of the nBT062-SMCC-DM1 (six doses of 13.8 μg/kg) completely blocked tumor growth during the dosing period [2]. |
| Enzyme Assay | Kadcyla® (T-DM1), an antibody-drug conjugates (ADCs) for HER2+ breast cancer treatment, has been approved by the Food and Drug Administration (FDA) in 2013. An ADC of random lysine conjugation, it has difficulties in DAR control and unsatisfactory PK due to uneven DAR distribution. It also gives rise to aggregation during conjugation because of the hydrophobicity nature of the cytotoxin, DM1. The linker-drug in T-DM1, SMCC-DM1 is hydrophobic and requires certain percentage of organic solvent such as DMA in the conjugation solution, limiting the manufacturing process in an organic-solvent-compatible device and adding extra costs. To address these problems, a site-specific conjugation method was developed involving full reduction of antibody and full conjugation with the bridge-like conjugator-drug, based on the work of Caddick and co-workers, to obtain a site-directed antibody-drug conjugate with DAR 4. The bridge-like conjugator was assembled with SMCC-DM1 and different lengths of hydrophilic polyethylene glycol (PEG) moiety. By applying PEG moiety in the side chain of the linker-drug, the organic solvent used in the conjugation can be reduced. When the PEG length is about 26 units, organic solvent is no longer needed in the conjugation. Reducing the amount of organic solvent in conjugation could also diminish the aggregation occurrence during the conjugation. Moreover, the conjugation configuration with the designed conjugator was also discussed in the article. The binding affinity of the resulting ADCs did not show significant decrease and the cell based assay and animal study have shown the comparable results with T-DM1.[1] |
| Cell Assay |
Growth inhibition assay and proliferation assay. The growth inhibitory effect of nBT062-SMCC-DM1, nBT062-SPDB-DM4, nBT062-SPP-DM1, and dexamethasone on growth of MM cell lines, PBMCs, and BM stromal cells (BMSC) was assessed by measuring 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide dye absorbance, as previously described (16). One antibody molecule has attached to it ∼3.5 molecules DM4. The molecular weight of the antibody is not significantly increased by the attachment of the DM4 molecules.[2] The ability for nBT062-SPDB-DM4 to mediate antigen-dependent bystander killing of proximal CD138-negative cells was evaluated. CD138-positive MM OPM2 cells (1 × 10-4 per well) and CD138-negative Namalwa cells (3 × 10-3 per well) were plated separately or mixed in 96-well round-bottomed plates and exposed to nBT062-SPDB-DM4 for 120 h. Cell viability was then assessed using WST-8 reagent. To evaluate growth inhibitory effects of immunoconjugates against MM cells in the BM milieu, MM cells (2 × 104 per well) were cultured for 48 h in BMSC (1 × 104 per well) coated 96-well plates (Costar) in the presence or absence of the drugs. DNA synthesis was measured by [3H]thymidine uptake, with [3H]thymidine (0.5 μCi/well) added during the last 8 h of 48-h cultures. All experiments were done in quadruplicate.[2] Cell cycle analysis. MM cells (1 × 106) were incubated with or without agents, washed with PBS, permeabilized by a 30-min exposure to 70% ethanol at −20°C, incubated with propidium iodide (50 μg/mL) in 0.5 mL PBS containing 20 units/mL RNase A for 30 min at room temperature, and analyzed for DNA content by using flow cytometry. |
| Animal Protocol |
Green fluorescent protein–positive human MM xenograft mouse model and SCID-hu mouse model.[2] OPM1 cells were transfected with green fluorescent protein (OPM1GFP+) using a lentiviral vector, as previously described. CB17 SCID mice (48-54 days old) were purchased from Charles River Laboratories. All animal studies were conducted according to protocols approved by the Animal Ethics Committee of the Dana-Farber Cancer Institute. Mice were inoculated s.c. with 5 × 106 OPM1GFP+ MM cells in 100 μL RPMI 1640. When tumors became palpable, mice were assigned into the treatment group receiving 200 μg conjugate per mouse via tail vein injection weekly or the control group receiving vehicle alone. Caliper measurements of the longest perpendicular tumor diameters were done every alternate day to estimate the tumor volume using the following formula representing the three-dimensional volume of an ellipse: 4 / 3 × (width / 2)2 × (length / 2). Animals were sacrificed when tumors reached 2 cm or when moribund. Survival was evaluated from the first day of treatment until death. Tumor growth was evaluated using caliper measurements from the first day of treatment until day of sacrifice, day 10 for control, and day 21 for the nBT062-SPDB-DM4 treatment group. Mice were monitored by whole-body fluorescence imaging using Illumatool Bright Light System LT-9900 (Lightools Research) after shaving the tumor area. The images were captured with a Canon IXY digital 700 camera. Ex vivo analysis of tumor image was captured with a LEICA DM IL microscope connected to the LEICA DFC300 FX camera at 40 units/0.60. Human fetal long bones were implanted into CB17 SCID mice (SCID-hu), as previously described (18). Briefly, 4 wk after bone implantation, 2.5 × 106 INA-6 cells in a final volume of 100 μL of RPMI 1640 were injected directly into the human BM cavity in the SCID-hu mice. An increase in the levels of soluble human IL-6 receptor (shuIL-6R), which is released by INA-6 cells, was used as a parameter of MM cell growth and burden of disease in SCID-hu mice. Mice developed measurable serum shuIL-6R ∼4 wk after INA-6 cell injection and then received 0.176 mg conjugate or vehicle control via tail vein injection weekly for 7 wk. After treatments, blood samples were collected and assayed for shuIL-6R levels by an ELISA. |
| References |
[1]. Site-specific and hydrophilic ADCs through disulfide-bridged linker and branched PEG. Bioorg Med Chem Lett. 2018 May 1;28(8):1363-1370. [2]. The monoclonal antibody nBT062 conjugated to cytotoxic Maytansinoids has selective cytotoxicity against CD138-positive multiple myeloma cells in vitro and in vivo. Clin Cancer Res. 2009 Jun 15;15(12):4028-37. doi: 10.1158/1078-0432.CCR-08-2867. [3]. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006 Apr 15;66(8):4426-33. |
| Additional Infomation |
Purpose: We investigated the antitumor effect of murine/human chimeric CD138-specific monoclonal antibody nBT062 conjugated with highly cytotoxic maytansinoid derivatives against multiple myeloma (MM) cells in vitro and in vivo.
Experimental design: We examined the growth inhibitory effect of BT062-SPDB-DM4, BT062-SMCC-DM1, and BT062-SPP-DM1 against MM cell lines and primary tumor cells from MM patients. We also examined in vivo activity of these agents in murine MM cell xenograft model of human and severe combined immunodeficient (SCID) mice bearing implant bone chips injected with human MM cells (SCID-hu model).
Results: Anti-CD138 immunoconjugates significantly inhibited growth of MM cell lines and primary tumor cells from MM patients without cytotoxicity against peripheral blood mononuclear cells from healthy volunteers. In MM cells, they induced G(2)-M cell cycle arrest, followed by apoptosis associated with cleavage of caspase-3, caspase-8, caspase-9, and poly(ADP-ribose) polymerase. Nonconjugated nBT062 completely blocked cytotoxicity induced by nBT062-maytansinoid conjugate, confirming that specific binding is required for inducing cytotoxicity. Moreover, nBT062-maytansinoid conjugates blocked adhesion of MM cells to bone marrow stromal cells. The coculture of MM cells with bone marrow stromal cells protects against dexamethasone-induced death but had no effect on the cytotoxicity of immunoconjugates. Importantly, nBT062-SPDB-DM4 and nBT062-SPP-DM1 significantly inhibited MM tumor growth in vivo and prolonged host survival in both the xenograft mouse models of human MM and SCID-hu mouse model.
Conclusion: These results provide the preclinical framework supporting evaluation of nBT062-maytansinoid derivatives in clinical trials to improve patient outcome in MM.[2] Researchers report that the huC242-maytansinoid conjugate, huC242-SMCC-DM1, with a “noncleavable” linker containing a thioether bond was at least as potent in vitro as the selected conjugate, huC242-SPDB-DM4, which has a “cleavable” linker containing a disulfide bond. This was surprising because huC242-SMCC-DM1 displayed significantly lower in vivo activity in multiple xenograft tumor models. To investigate this conundrum, we undertook a series of experiments to elucidate the mechanism of cell killing by the conjugates. The results delineate an activation process, for both conjugates, that requires lysosomal degradation of the antibody component of the conjugate. However, distinct maytansinoid metabolites produced by intracellular processing of huC242-SPDB-DM4 were identified and characterized, providing a likely mechanism for its superior antitumor efficacy.[3] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~16.67 mg/mL (~15.54 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: 2 mg/mL (1.86 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution; with sonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2 mg/mL (1.86 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2 mg/mL (1.86 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 10% DMSO+ 40% PEG300+ 5% Tween-80+ 45% saline: 2 mg/mL (1.86 mM) (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 0.9323 mL | 4.6615 mL | 9.3231 mL | |
| 5 mM | 0.1865 mL | 0.9323 mL | 1.8646 mL | |
| 10 mM | 0.0932 mL | 0.4662 mL | 0.9323 mL |