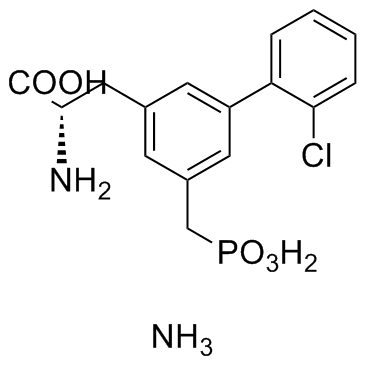
SDZ 220-581 Ammonium salt (SDZ-220581; SDZ220581) is a novel and competitive antagonist of NMDA glutamate receptor subtype (pKi= 7.7) with the potential to be used for Parkinson's disease.
Physicochemical Properties
| Molecular Formula | C16H20CLN2O5P |
| Molecular Weight | 386.767164230347 |
| Exact Mass | 386.079 |
| CAS # | 179411-94-0 |
| Related CAS # | SDZ 220-581;174575-17-8;SDZ 220-581 hydrochloride;179411-93-9 |
| PubChem CID | 74892038 |
| Appearance | Off-white to light yellow solid powder |
| Hydrogen Bond Donor Count | 5 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 25 |
| Complexity | 485 |
| Defined Atom Stereocenter Count | 1 |
| SMILES | ClC1=CC=CC=C1C2=CC(C[C@@H](C(O)=O)N)=CC(CP(O)(O)=O)=C2.N |
| InChi Key | JSKZYMJZKPLCNJ-RSAXXLAASA-N |
| InChi Code | InChI=1S/C16H17ClNO5P.H3N/c17-14-4-2-1-3-13(14)12-6-10(8-15(18)16(19)20)5-11(7-12)9-24(21,22)23;/h1-7,15H,8-9,18H2,(H,19,20)(H2,21,22,23);1H3/t15-;/m0./s1 |
| Chemical Name | (2S)-2-amino-3-[3-(2-chlorophenyl)-5-(phosphonomethyl)phenyl]propanoic acid;azane |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| ln Vitro |
Inhibits the mucosal transport of [¹⁴C]L-phenylalanine in isolated rat jejunal brush border membrane vesicles. At a concentration of 50 μM, SDZ EAB 515 (the lead compound in the series) significantly inhibited transport, suggesting interaction with the large neutral amino acid carrier system. SDZ 220-581 belongs to this chemical series. [1] |
| ln Vivo |
Male OF-1 mice were dose-dependently protected against maximal electroshock epileptic seizures (MES) by treatment with SDZ 220-581 (3.2–32 mg/kg; p.o.; for 24 hours). The quick onset and extended duration of action of SDZ 220-581 are characteristics of its protective duration [1]. Anticonvulsant Activity: Orally administered SDZ 220-581 dose-dependently protected mice and rats against maximal electroshock-induced seizures (MES). In mice, the ED₅₀ was <3.2 mg/kg p.o., with a rapid onset (≤1 hr) and long duration (≥24 hr) of action. Full protection in rats was achieved at 10 mg/kg p.o. [1] Neuroprotective Activity (Quinolinic Acid Model): SDZ 220-581 reduced the size of striatal lesions induced by intrastriatal quinolinic acid injection in rats. Protection was observed after both intraperitoneal (3-15 mg/kg) and oral (10-50 mg/kg) administration, as assessed by magnetic resonance imaging (MRI) and preservation of choline acetyltransferase (CAT) and glutamate decarboxylase (GAD) activities. [1] Neuroprotective Activity (Ischemia Model): In a rat model of permanent middle cerebral artery occlusion (MCAO), a single intravenous bolus of SDZ 220-581 (1.25 mg/kg) administered 15 minutes before occlusion reduced cerebral infarct size by 40% (measured by MRI) and by approximately 30% (measured by TTC staining). A U-shaped dose-response was noted. Oral administration (10 and 30 mg/kg, twice) 18 and 1 hour before MCAO also reduced infarct size by 20-30%. [1] Analgesic Activity: SDZ 220-581 showed analgesic effects in models of neuropathic and inflammatory pain. Oral administration significantly reversed mechanical hyperalgesia in a rat model of partial sciatic nerve ligation at low doses. It also produced a dose-related reversal of Freund's adjuvant-induced mechanical hyperalgesia in the rat knee (3-30 mg/kg p.o.). [1] Effect on 2-Deoxyglucose Uptake: Intraperitoneal administration of SDZ 220-581 (6-10 mg/kg) altered regional cerebral glucose utilization in rats, characterized by increased uptake in parts of the extrapyramidal and limbic systems and decreased uptake in some sensory areas. [1] Interaction with L-DOPA: Subcutaneous administration of low doses (≥0.05 mg/kg) of SDZ 220-581 counteracted the antiparkinsonian effects (reversal of motor deficits and increased locomotor activity) of L-DOPA in MPTP-treated marmosets. [1] Lack of Tolerance and Receptor Upregulation: Subchronic oral administration (10 mg/kg/day for 10 days) of SDZ 220-581 did not reduce its neuroprotective efficacy against quinolinic acid toxicity. Chronic treatment did not alter NMDA receptor density or affinity in various brain regions as measured by [³H]CGP-39653 binding. [1] |
| Animal Protocol |
Animal/Disease Models: Male OF-1 mouse (18-26g) [1] Doses: 3.2mg/kg, 10mg/kg, 32mg/kg Route of Administration: Oral administration; Route of Administration: Oral administration. 24-hour Experimental Results: Dose-dependent protection of mice from maximal electroshockable seizures (MES) following oral administration. Maximal Electroshock Seizure (MES) in Mice/Rats: Male OF-1 mice or Sprague-Dawley rats were used. Tonic hind-limb convulsions were induced via corneal electrodes using constant current pulses. Test compounds were administered orally. Animals not showing tonic hind-limb extension were considered protected. Protection was assessed at various time points post-administration. [1] Rotarod Performance (Motor Incapacitation): Mice or rats were placed on a rotating rod. The time they remained on the rod before falling was measured up to a cutoff of 120 seconds. Readings were taken at intervals after oral drug administration. [1] Quinolinic Acid-Induced Striatal Lesion: Under anesthesia, quinolinic acid (250 nmoles) was injected into the right striatum of male Sprague-Dawley rats. Test compounds were administered intraperitoneally or orally as specified. Lesion size was assessed 24 hours later by T2-weighted magnetic resonance imaging (MRI) in vivo, or 7 days later by post-mortem measurement of striatal choline acetyltransferase (CAT) and glutamate decarboxylase (GAD) activities. [1] Middle Cerebral Artery Occlusion (MCAO): Under anesthesia, the left middle cerebral artery of Sprague-Dawley or Fischer 344 rats was permanently occluded. Test compounds were administered intravenously (as a single bolus before or after occlusion) or orally (twice before occlusion). Infarct size was determined 24 hours post-occlusion by MRI or by histological analysis using 2,3,5-triphenyl tetrazolium chloride (TTC) staining 4 days post-occlusion. Neurological status was also evaluated. [1] Neuropathic Pain Model (Partial Sciatic Nerve Ligation): In male Sprague-Dawley rats, the left sciatic nerve was partially ligated. Mechanical hyperalgesia in the ipsilateral foot was assessed 12-15 days post-ligation using a Randall-Selitto paw pressure apparatus. Drugs were administered orally, and withdrawal thresholds were measured. [1] Inflammatory Pain Model (Freund's Adjuvant): Freund's complete adjuvant was injected into one knee of female Sprague-Dawley rats. Three days later, mechanical hyperalgesia was assessed by measuring the load tolerance on the injected vs. uninjected leg. Drugs were administered orally. [1] 2-Deoxyglucose Uptake Study: Male Wistar rats received an intraperitoneal injection of the test compound. One hour later, [¹⁴C]2-deoxyglucose was injected intravenously. After 45 minutes, animals were sacrificed, brains were sectioned, and autoradiograms were prepared and quantified to measure relative regional cerebral glucose uptake. [1] MPTP-Treated Marmoset Model: Common marmosets previously treated with MPTP to induce stable motor deficits were used. On test days, after acclimatization in activity cages, they received carbidopa (12.5 mg/kg p.o.), followed 60 minutes later by L-DOPA (2.5 mg/kg p.o.) alone or in combination with subcutaneous SDZ 220-581. Locomotor activity was monitored automatically, and motor disability was scored by an observer. [1] Brain Uptake Index (BUI) Measurement: Under anesthesia, a bolus containing [¹⁴C]L-phenylalanine and [³H]H₂O, with or without unlabeled inhibitor, was rapidly injected into the common carotid artery of male Wistar rats. Animals were decapitated 15 seconds later. Radioactivity in the injected hemisphere and injection solution was measured to calculate the Brain Uptake Index. [1] |
| ADME/Pharmacokinetics |
SDZ 220-581 exhibits good oral bioavailability and brain penetration, attributed possibly to its lipophilicity and potential interaction with the large neutral amino acid carrier system. It has a long duration of action; compound was detectable in blood plasma 24 hours after oral administration to rats. The L-enantiomers of this compound class may utilize carriers for active transport from the intestine and/or through the blood-brain barrier. [1] |
| Toxicity/Toxicokinetics |
Motor Impairment: In rotarod tests, oral doses of SDZ 220-581 that significantly impaired performance in mice (32 and 100 mg/kg) were about 10 times higher than the fully protective dose against MES (10 mg/kg). In rats, 100 mg/kg p.o. caused only marginal impairment, while 300 mg/kg p.o. caused complete incapacitation. [1] Psychotomimetic Potential: The potential for psychotomimetic side-effects (e.g., hallucinations) common to NMDA antagonists is noted, but the "atypical" interaction profile of SDZ 220-581 with dopaminergic systems might indicate a different side-effect potential. The risk may be lower with oral vs. intravenous administration. [1] Neurotoxicity (Vacuolization): High intravenous doses (20 mg/kg i.v., but not 6 mg/kg i.v.) of SDZ 220-581 induced vacuolization of cortical neurons in rats, a phenomenon observed with other NMDA antagonists. Its occurrence at therapeutically relevant oral doses was not determined. [1] |
| References |
[1]. Urwyler S, et al. Biphenyl-derivatives of 2-amino-7-phosphono-heptanoic acid, a novel class of potent competitive N-methyl-D-aspartate receptor antagonists--II. Pharmacological characterization in vivo. Neuropharmacology. 1996 Jun;35(6):655-69. [2]. Gilmour G, et al. In vitro characterisation of the novel positive allosteric modulators of the mGlu₅ receptor, LSN2463359 and LSN2814617, and their effects on sleep architecture and operant responding in the rat. Neuropharmacology. 2013 Jan;64:224-39. |
| Additional Infomation |
SDZ 220-581 is a biphenyl derivative of 2-amino-7-phosphonoheptanoic acid (AP7). It represents a novel class of competitive NMDA receptor antagonists with high oral efficacy and a long duration of action, potentially allowing once-daily administration. Its pharmacological profile differs from earlier antagonists in aspects such as effects on cerebral glucose uptake patterns and interaction with dopaminergic therapies. It is suggested for potential therapeutic applications in epilepsy, neuroprotection (e.g., stroke, neurodegenerative diseases), neuropathic pain, and possibly drug abuse/withdrawal, but not in Parkinson's disease due to its antagonism of L-DOPA effects. [1] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5855 mL | 12.9276 mL | 25.8552 mL | |
| 5 mM | 0.5171 mL | 2.5855 mL | 5.1710 mL | |
| 10 mM | 0.2586 mL | 1.2928 mL | 2.5855 mL |