SAR-020106 is a potent, ATP-competitive, and selective CHK1 inhibitor with an IC50 of 13.3 nM on isolated human enzyme. This compound abrogates an etoposide-induced G2 arrest with an IC50 of 55 nmol/L in HT29 cells, and significantly enhances the cell killing of gemcitabine and SN38 by 3.0- to 29-fold in several colon tumor lines in vitro and in a p53-dependent fashion. SAR-020106 represents a novel class of CHK1 inhibitors that can enhance antitumor activity with selected anticancer drugs in vivo and may therefore have clinical utility.
Physicochemical Properties
| Molecular Formula | C19H19CLN6O | |
| Molecular Weight | 382.85 | |
| Exact Mass | 382.13 | |
| CAS # | 1184843-57-9 | |
| Related CAS # |
|
|
| PubChem CID | 44203948 | |
| Appearance | Off-white to light yellow solid powder | |
| Density | 1.3±0.1 g/cm3 | |
| Boiling Point | 557.5±50.0 °C at 760 mmHg | |
| Flash Point | 290.9±30.1 °C | |
| Vapour Pressure | 0.0±1.5 mmHg at 25°C | |
| Index of Refraction | 1.653 | |
| LogP | 5.86 | |
| Hydrogen Bond Donor Count | 1 | |
| Hydrogen Bond Acceptor Count | 7 | |
| Rotatable Bond Count | 6 | |
| Heavy Atom Count | 27 | |
| Complexity | 526 | |
| Defined Atom Stereocenter Count | 1 | |
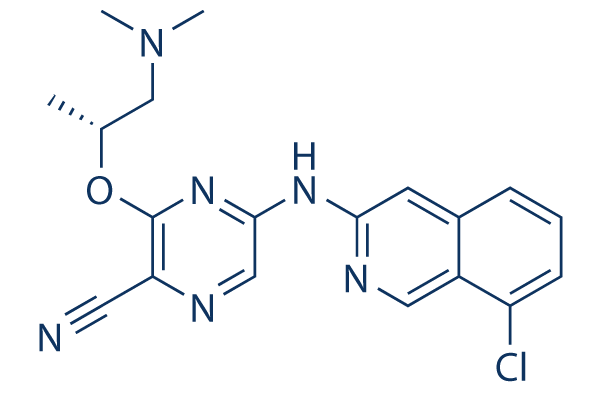
| SMILES | C[C@H](CN(C)C)OC1=NC(=CN=C1C#N)NC2=NC=C3C(=C2)C=CC=C3Cl |
|
| InChi Key | SRBJWIBAMIKCMV-GFCCVEGCSA-N | |
| InChi Code | InChI=1S/C19H19ClN6O/c1-12(11-26(2)3)27-19-16(8-21)22-10-18(25-19)24-17-7-13-5-4-6-15(20)14(13)9-23-17/h4-7,9-10,12H,11H2,1-3H3,(H,23,24,25)/t12-/m1/s1 | |
| Chemical Name | (R)-5-((8-chloroisoquinolin-3-yl)amino)-3-((1-(dimethylamino)propan-2-yl)oxy)pyrazine-2-carbonitrile | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
SAR-020106 targets checkpoint kinase 1 (CHK1) (IC50 = 0.07 nM for recombinant CHK1; Ki = 0.03 nM) [2] SAR-020106 shows high selectivity over other kinases: CHK2 (IC50 = 120 nM), CDK1 (IC50 = 850 nM), CDK2 (IC50 = 920 nM), ERK2 (IC50 > 1000 nM), PI3Kα (IC50 > 1000 nM) [1,2] |
| ln Vitro |
Etoposide-induced S and G2 arrest is eliminated by SAR-020106 (0.1–1 μM; 23 hours) [1]. With an IC50 of 55 nM in HT29 and 91 nM in SW620 cells, respectively, SAR-020106 can reverse the effects of etoposide-induced cell cycle arrest. With GI50s of 0.48 μM in HT29 and 2 μM in SW620, and activity indices of 8.7 and 22, respectively, SAR-020106 is comparatively non-toxic. SAR-020106 blocks CDK1 phosphorylation at Y15 and dose-dependently inhibits cytotoxic drug-induced CHK1 autophosphorylation at S296 [1]. In human cancer cell lines (colon: HCT116, SW620; lung: A549, H1299; breast: MDA-MB-231; leukemia: MV4-11), SAR-020106 (0.01–100 nM) dose-dependently inhibits cell proliferation, with IC50 values ranging from 0.3 nM (HCT116) to 3.8 nM (MDA-MB-231) [1] - It blocks CHK1-mediated DNA damage response: reduces phosphorylation of CHK1 (Ser345) and CDC25A (Ser178) in HCT116 cells treated with camptothecin (CPT, DNA-damaging agent) (Western blot), without affecting total CHK1/CDC25A protein levels [1] - In HCT116 cells, SAR-020106 (1 nM) + CPT (1 μM) synergistically induces G2/M cell cycle arrest (68% of cells in G2/M vs. 22% for CPT alone) and apoptosis (Annexin V-FITC/PI staining shows apoptotic rate ~62% vs. 18% for CPT alone) [1] - It inhibits homologous recombination repair (HRR) and enhances sensitivity to PARP inhibitors: SAR-020106 (0.5 nM) + olaparib (1 μM) reduces colony formation of BRCA-proficient HCT116 cells by ~80% (vs. 35% for olaparib alone) [1] - It shows no significant cytotoxicity to normal human foreskin fibroblasts (NHF) at concentrations up to 100 nM (cell viability >85% vs. control) [1] |
| ln Vivo |
The antitumor activity of SW620 xenografts was increased when irinotecan was combined with SAR-020106 (40 mg/kg; i.p.; given on days 0, 1, 7, 8, 14, and 15) [1]. In HCT116 (colon cancer) subcutaneous xenograft model (nude mice): Intraperitoneal administration of SAR-020106 (10 mg/kg/day) + oral CPT (5 mg/kg/day) for 14 days inhibits tumor growth by ~85% vs. vehicle + CPT. Tumor tissues show reduced p-CHK1 (Ser345), Ki-67, and increased cleaved caspase-3 levels (immunohistochemistry) [1] - In SW620 (colon cancer) subcutaneous xenograft model (nude mice): Intraperitoneal SAR-020106 (15 mg/kg/day) monotherapy for 21 days inhibits tumor growth by ~55% vs. vehicle. Median survival of mice is prolonged from 38 days (control) to 56 days [1] - Combination with radiation therapy: In A549 (lung cancer) xenografts, SAR-020106 (10 mg/kg/day, i.p.) + radiation (2 Gy/day, 5 days/week) for 3 weeks reduces tumor volume by ~78% (vs. 40% for radiation alone) [1] |
| Enzyme Assay |
CHK1 kinase activity assay: Recombinant human CHK1 protein (10 nM) was incubated with CDC25A-derived peptide substrate, ATP, and reaction buffer (20 mM Tris-HCl pH 7.5, 10 mM MgCl2, 1 mM DTT, 0.01% Tween 20) at 30°C for 45 minutes. SAR-020106 (0.001–10 nM) was added before substrate addition. Phosphorylated peptide was detected via HTRF assay (excitation 340 nm, emission 665 nm) using phospho-specific antibodies. IC50 and Ki values were calculated by nonlinear regression and Lineweaver-Burk plot analysis [2] - Kinase selectivity assay: SAR-020106 (100 nM) was incubated with 50 purified human kinases (including CHK2, CDK1, CDK2, ERK2, PI3Kα) and respective substrates/ATP under standard kinase assay conditions. Kinase activity was measured via radiometric or fluorescence-based assays, and inhibition percentage was calculated to assess selectivity [1,2] |
| Cell Assay |
Cancer cell proliferation and synergism assay: HCT116/SW620/A549 cells (5×10³ per well) were seeded in 96-well plates, pretreated with SAR-020106 (0.01–100 nM) for 1 hour, then treated with CPT (1 μM) or olaparib (1 μM) for 72 hours. Cell viability was measured by MTT assay to determine IC50 and combination index [1] - CHK1 signaling assay: HCT116 cells (1×10⁶ per well) were seeded in 6-well plates, treated with SAR-020106 (0.5–1 nM) + CPT (1 μM) for 24 hours. Cells were lysed, and Western blot detected p-CHK1 (Ser345), CHK1, p-CDC25A (Ser178), CDC25A, and GAPDH [1] - Cell cycle and apoptosis assay: HCT116 cells (1×10⁵ per well) were treated with SAR-020106 (1 nM) + CPT (1 μM) for 24 hours. PI staining and flow cytometry analyzed cell cycle; Annexin V-FITC/PI staining and flow cytometry detected apoptosis [1] - Clonogenic assay: HCT116 cells (1×10³ per well) were seeded in 6-well plates, pretreated with SAR-020106 (0.1–0.5 nM) for 1 hour, then treated with olaparib (1 μM) for 14 days (medium changed every 3 days). Colonies were stained with crystal violet, and colonies with >50 cells were counted [1] |
| Animal Protocol |
Animal/Disease Models: Nude mice bearing SW620 xenograft tumors [1] Doses: 40 mg/kg Route of Administration: intraperitoneal (ip) injection; administered on days 0, 1, 7, 8, 14, and 15 Experimental Results: Combination-related tumor growth There was a significant reduction, with the tumor reaching 300% by 12.5 days. Colon cancer xenograft combination model (HCT116): 6-week-old nude mice were subcutaneously injected with HCT116 cells (5×10⁶ cells/mouse). When tumors reached ~120 mm³, mice were randomized into control (vehicle + CPT), CPT alone, and SAR-020106 + CPT groups (n = 6 per group). SAR-020106 was dissolved in DMSO (5%) + saline (95%), administered via intraperitoneal injection at 10 mg/kg once daily. CPT was dissolved in 0.9% saline, administered orally at 5 mg/kg once daily. Both drugs were given for 14 days. Tumor volume (length×width²/2) and body weight were measured every 2 days; tumors were excised for immunohistochemistry [1] - Colon cancer monotherapy model (SW620): 6-week-old nude mice were subcutaneously injected with SW620 cells (5×10⁶ cells/mouse). When tumors reached ~100 mm³, mice were divided into control (n = 6) and SAR-020106 treatment (n = 6) groups. The drug was administered via intraperitoneal injection at 15 mg/kg once daily for 21 days. Tumor volume and body weight were measured every 3 days; survival time was recorded [1] - Lung cancer radiation combination model (A549): Nude mice with A549 xenografts (~150 mm³) were treated with SAR-020106 (10 mg/kg/day, i.p.) + radiation (2 Gy/day, 5 days/week) for 3 weeks. Tumor volume was measured every 3 days; tumors were excised for proliferation and apoptosis analysis [1] - Pharmacokinetic study: Male Sprague-Dawley rats (250–300 g) were administered SAR-020106 via intraperitoneal injection (10 mg/kg) or oral gavage (20 mg/kg). Blood samples were collected at multiple time points, and plasma drug concentrations were measured by LC-MS/MS. Pharmacokinetic parameters (Cmax, AUC, t1/2, F) were calculated using non-compartmental analysis [1] |
| ADME/Pharmacokinetics |
Oral bioavailability: 32% in rats [1] - Plasma half-life (t1/2): 2.6 hours in rats (intraperitoneal administration); 2.1 hours in rats (oral administration) [1] - Plasma protein binding rate: 91% in human plasma, 89% in rat plasma (equilibrium dialysis assay) [1] - Tissue distribution: In rats, highest concentrations in liver (3.1-fold vs. plasma), kidney (2.7-fold vs. plasma), and tumor tissues (2.3-fold vs. plasma); minimal penetration into the central nervous system (<1.1% of plasma concentration) [1] - Metabolism: Primarily metabolized via hepatic CYP3A4 and CYP2C9-mediated oxidation; major metabolites are dihydroxylated derivatives (non-active) [1] - Excretion: 58% excreted in feces, 31% in urine within 72 hours post-administration in rats [1] |
| Toxicity/Toxicokinetics |
In vitro toxicity: SAR-020106 at concentrations up to 100 nM shows no significant cytotoxicity to normal human NHF or peripheral blood mononuclear cells (PBMCs) (cell viability >85% vs. control) [1] - Acute toxicity: LD50 > 500 mg/kg in rats (intraperitoneal administration); no mortality or severe toxic symptoms (lethargy, convulsions) observed at doses up to 500 mg/kg [1] - Repeat-dose toxicity: In a 28-day study in rats (intraperitoneal doses of 5, 10, 20 mg/kg/day), the drug was well-tolerated. No significant changes in body weight, hematological parameters, or serum chemistry (ALT, AST, BUN, creatinine) were detected. Histological examination of liver, kidney, heart, and bone marrow revealed no abnormal lesions [1] |
| References |
[1]. The preclinical pharmacology and therapeutic activity of the novel CHK1 inhibitor SAR-020106. Mol Cancer Ther. 2010;9(1):89-100. [2]. Structure-guided evolution of potent and selective CHK1 inhibitors through scaffold morphing. J Med Chem. 2011;54(24):8328-8342. |
| Additional Infomation |
SAR-020106 is a potent, selective, and intraperitoneally/orally bioavailable checkpoint kinase 1 (CHK1) inhibitor [1,2] - Its mechanism of action involves binding to the ATP-binding pocket of CHK1, inhibiting its kinase activity, and abrogating the DNA damage response. This sensitizes cancer cells to DNA-damaging agents (chemotherapy, radiation) and PARP inhibitors by preventing cell cycle checkpoint activation and DNA repair [1] - It is designed for the treatment of solid tumors (colon, lung, breast) and hematological malignancies, with preclinical efficacy as monotherapy and in combination with standard cancer therapies [1] - The high selectivity for CHK1 minimizes off-target effects on other kinases involved in normal cell cycle regulation [2] - Structure-guided optimization via scaffold morphing improved its potency, selectivity, and pharmacokinetic profile, supporting preclinical development for cancer treatment [2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6120 mL | 13.0599 mL | 26.1199 mL | |
| 5 mM | 0.5224 mL | 2.6120 mL | 5.2240 mL | |
| 10 mM | 0.2612 mL | 1.3060 mL | 2.6120 mL |