S-2101 is a novel and potent lysine-specific demethylase 1 (LSD1) inhibitor with anticancer activity. It inhibits LSD1 with an IC50 of 0.99 μM, Ki of 0.61 μM and Kinact/Ki of 4560 M/s.
Physicochemical Properties
| Exact Mass | 311.09 |
| Elemental Analysis | C, 61.64; H, 5.17; Cl, 11.37; F, 12.19; N, 4.49; O, 5.13 |
| CAS # | 1239262-36-2 |
| PubChem CID | 121513887 |
| Appearance | White to off-white solid powder |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 4 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 21 |
| Complexity | 322 |
| Defined Atom Stereocenter Count | 2 |
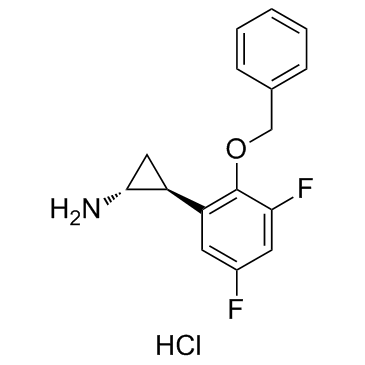
| SMILES | C1[C@H]([C@@H]1N)C2=C(C(=CC(=C2)F)F)OCC3=CC=CC=C3.Cl |
| InChi Key | CTSUZAFTJVLOIX-SBKWZQTDSA-N |
| InChi Code | InChI=1S/C16H15F2NO.ClH/c17-11-6-13(12-8-15(12)19)16(14(18)7-11)20-9-10-4-2-1-3-5-10;/h1-7,12,15H,8-9,19H2;1H/t12-,15+;/m0./s1 |
| Chemical Name | (1R,2S)-2-(3,5-difluoro-2-phenylmethoxyphenyl)cyclopropan-1-amine;hydrochloride |
| Synonyms | S2101; S-2101; S 2101 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | KDM1/lysine-specific demethylase 1 (LSD1) |
| ln Vitro | With an IC50 of 0.99 μM, Ki of 0.61 μM, and Kinact/Ki of 4560 M/s, S2101 is an inhibitor of lysine-specific demethylase 1 (LSD1). Additionally, S2101 has significantly less inhibitory activity in response to MAO-A (Ki=110 µM, Kinact/Ki=60 M/s) and MAO-B (Ki=17 µM, Kinact/Ki=18 M/s). H3K4me2 is treated with S2101 in HEK293T cells, and this treatment causes a dose-dependent rise in H3K4me2, which must have accumulated due to LSD1 inactivation. The levels of LSD1 and histone H3 in the nuclear extracts do not significantly change during the S2101 therapy. Since 50 μM 2-PCPA elicits a similar amount of H3K4me2 when treated with 1 μM S2101, it is assumed that S2101 has roughly 50-fold greater LSD1 inhibition action than 2-PCPA in human cells[1]. |
| Enzyme Assay |
LSD1 Inhibition Assays[1] The kinetic inhibition parameters of LSD1 demethylase inhibition were obtained using the peroxidase-coupled reaction method. The LSD1 protein (3.0 pmol) was incubated with 40 μM H3K4me2 peptides, in a buffer containing 10 mM HEPES-Na (pH 7.5), 200 μM 4-aminoantipyrine, the Modified Trinder’s reagent TOOS [N-ethyl-N-(2-hydroxy-3-sulfopropyl)-3-methylaniline, sodium salt, dihydrate], and 0.01 mg/mL horseradish peroxidase, for 10 min. This system generates a kcat of 7.61 ± 0.56 min−1 and a Km of 16 ± 5 μM. The horseradish peroxidase converts TOOS and 4-aminoantipyrine, using the peroxidase byproduct produced by LSD1 demethylation, to a pink/violet-colored compound, with a λmax of 555 nm (http://www.dojindo.co.jp/protocol/protocol/trinder.pdf). The reaction mixture was prepared in the dark to inhibit the reactivity of the Trinder’s reagent. The absorption of the reaction mixture was measured at 562 nm every 10 s with a microplate reader (Ultrospec Visible Plate Reader II 96, GE Healthcare). Although the λmax for TOOS is 555 nm, measuring the wavelength at 562 nm was sufficient to provide accurate results of the inhibition, since the equations used to calculate KI are dependent on the difference in activity, and not the absolute values. The results were transformed to the amounts of demethylated peptides for LSD1, and oxidized tyramine for MAOs, using the ε value for TOOS of 3.92 × 104. The results were analyzed and fit with the slow-binding inhibitor equation (eq 1) to yield kobs and were further transformed to calculate kinact and KIapp according to the Kitz−Wilson analysis, or with an offset value koffset when the y-intercept was clearly non-zero (eq 2) (26-31). KIapp was transformed to KI to exclude the bias from substrate concentrations (eq 3) (26). Data were fit with GraFit (http://www.erithacus.com/grafit/) and KyPlot (http://www.kyenslab.com/jp/). MAO Inhibition Assays[1] MAO inhibition assays were performed using the peroxidase-coupled reaction method, in essentially the same manner described for LSD1, but with 40 or 50 μM tyramine as the substrate and 9.0 and 4.5 μg of MAO-A and MAO-B, respectively. The results were analyzed and calculated in the same manner described for LSD1. Each result was calculated from at least three individual experiments. The symbols plotted on the time course assays are the averages of the experiments, and the best-fit line was calculated and fit by the slow-binding inhibitor equation, using KyPlot. |
| Cell Assay |
Western Blot Analysis of Compound-Treated Cells[1] HEK293T cells were grown in 100 mm dishes in Dulbecco’s modified Eagle’s medium, containing 10% FBS and a 100 units/mL penicillin, 100 μg/mL streptomycin antibiotic solution under a 5% CO2 atmosphere at 37 °C. Cells at approximately 70% confluency were treated with LSD1 inhibitors for 24 h, detached, and rinsed several times with ice-cold PBS. Nuclear extracts were prepared using a CelLytic NuCLEAR extraction kit, according to the manufacturer’s instructions. Each nuclear extract was electrophoresed on a 10 to 20% SDS−polyacrylamide gel and then transferred to a nitrocellulose membrane. The proteins on the membranes were probed with primary antibodies (anti-H3, Abcam ab1791; anti-H3K4me2, Upstate 07-030; and anti-LSD1) and then with a secondary antibody (goat anti-rabbit IgG−horseradish peroxidase conjugates) and were detected with a chemiluminescent system, Immobilon Western. |
| References |
[1]. Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1 . Biochemistry. 2010 Aug 3;49(30):6494-503. |
| Additional Infomation | Lysine-specific demethylase 1 (LSD1/KDM1) demethylates histone H3, in addition to tumor suppressor p53 and DNA methyltransferase 1 (Dnmt1), thus regulating eukaryotic gene expression by altering chromatin structure. Specific inhibitors of LSD1 are desired as anticancer agents, because LSD1 aberrations are associated with several cancers, and LSD1 inhibition restores the expression of abnormally silenced genes in cancerous cells. In this study, we designed and synthesized several candidate compounds to inhibit LSD1, based on the structures of LSD1 and monoamine oxidase B (MAO-B), in complex with an antidepressant tranylcypromine (2-PCPA) derivative. Compound S2101 exhibited stronger LSD1 inhibition than tranylcypromine and the known small LSD1 inhibitors in LSD1 demethylation assays, with a k(inact)/K(I) value of 4560 M(-1) s(-1). In comparison with tranylcypromine, the compound displayed weaker inhibition to the monoamine oxidases. The inhibition modes of the two 2-PCPA derivatives, 2-PFPA and S1201, were identified by determination of the inhibitor-bound LSD1 structures, which revealed the enhanced stability of the inhibitor-FAD adducts by their interactions with the surrounding LSD1 residues. These molecules are potential pharmaceutical candidates for cancer or latent virus infection, as well as research tools for LSD1-related biological investigations[1]. |
Solubility Data
| Solubility (In Vitro) | DMSO : ~100 mg/mL (~320.77 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: 5 mg/mL (16.04 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with sonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 50.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 5 mg/mL (16.04 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 50.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 5 mg/mL (16.04 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 50.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |