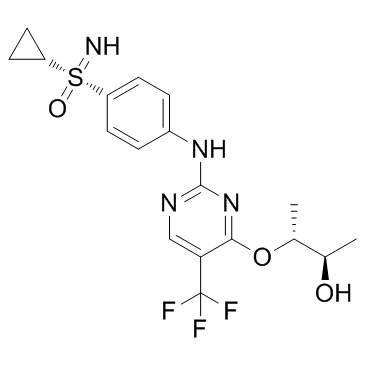
Roniciclib (formerly also known as BAY1000394; BAY-1000394) is a novel, potent and orally bioavailable pan-cyclin dependent kinase (CDK) inhibitor wwith anticancer activity. It inhibits CDK1, CDK2, CDK3, CDK4, CDK7, and CDK9 with IC50 values ranging from 5 to 25 nM. Because it specifically binds to and inhibits the activity of serine/threonine kinases involved in the regulation of cell cycle progression and cellular proliferation, namely CDK1/Cyclin B, CDK2/Cyclin E, CDK4/Cyclin D1, and CDK9/Cyclin T1, it demonstrates its antineoplastic activity. Apoptosis is induced and tumor cell proliferation is inhibited when these kinases are inhibited, which results in cell cycle arrest during the G1/S transition. Presently undergoing phase I clinical trials is BAY 1000394, a novel oral cytotoxic agent. The loss of cell-cycle checkpoint function and increased expression of antiapoptotic proteins resulting from deregulated activity of cyclin-dependent kinases (CDK) has been directly associated with the molecular pathology of cancer. With IC(50) values ranging from 5 to 25 nmol/L, BAY 1000394 suppresses the activity of transcriptional CDKs CDK7 and CDK9 as well as cell-cycle CDKs CDK1, CDK2, CDK3, and CDK4. In a variety of human cancer cell lines, cell proliferation was suppressed at low nanomolar concentrations. The suppression of phosphorylation of the CDK substrates RNA polymerase II, nucleophosmin, and retinoblastoma protein was demonstrated in assays involving cells. Cell-cycle profiles aligned with the suppression of CDK 1, 2, and 4, as demonstrated by cell-cycle block and release tests. BAY 1000394's physicochemical and pharmacokinetic characteristics enable quick absorption and a moderate oral bioavailability. After oral dosage, the substance significantly suppresses the growth of several human tumor xenografts on athymic mice, including models of chemotherapy resistance. Moreover, BAY 1000394 exhibits efficacy greater than additive when paired with etoposide and cisplatin.
Physicochemical Properties
| Molecular Formula | C18H21F3N4O3S |
| Molecular Weight | 430.44500 |
| Exact Mass | 430.129 |
| Elemental Analysis | C, 50.23; H, 4.92; F, 13.24; N, 13.02; O, 11.15; S, 7.45 |
| CAS # | 1223498-69-8 |
| Related CAS # | 1223498-69-8 |
| PubChem CID | 45380979 |
| Appearance | White to off-white solid powder |
| LogP | 4.993 |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 10 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 29 |
| Complexity | 661 |
| Defined Atom Stereocenter Count | 2 |
| SMILES | C[C@@H](O)[C@H](OC1=NC(NC2=CC=C([S@@](=N)(C3CC3)=O)C=C2)=NC=C1C(F)(F)F)C |
| InChi Key | UELYDGOOJPRWGF-MFOHZAOFSA-N |
| InChi Code | InChI=1S/C18H21F3N4O3S/c1-10(26)11(2)28-16-15(18(19,20)21)9-23-17(25-16)24-12-3-5-13(6-4-12)29(22,27)14-7-8-14/h3-6,9-11,14,22,26H,7-8H2,1-2H3,(H,23,24,25)/t10-,11-,29?/m1/s1 |
| Chemical Name | (2R,3R)-3-[2-[4-(cyclopropylsulfonimidoyl)anilino]-5-(trifluoromethyl)pyrimidin-4-yl]oxybutan-2-ol |
| Synonyms | Roniciclib; BAY1000394; roniciclib; 1223498-69-8; 0W9Q8U337A; BAY10-00394; BAY-1000,394; BAY-1000394; BAY 1000394 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Cdk1/cyclin B (IC50 = 7 nM); CDK2/cyclinE (IC50 = 9 nM); CDK4/cyclin D (IC50 = 11 nM); CDK9/cyclinT1 (IC50 = 5 nM); CDK7/Cyclin H/MAT1 (IC50 = 25 nM) Roniciclib (BAY-1000394; BAY1000394) inhibits the kinase activity of the cell-cycle CDKs CDK1/cyclin B, CDK2/cyclin E, and CDK4/cyclinDwith IC50 values of 7, 9, and 11 nM, respectively. The range of inhibition for the transcriptional CDKs CDK9/cyclin T1 and CDK7/cyclin H/MAT1 is comparable (5 and 25 nM)[1]. With a very balanced profile (mean IC50 on human tumor cells: 16 nM), riciclib potently inhibits the proliferation of several human and murine tumor cell lines[2]. Roniciclib (BAY 1000394) is a potent pan-cyclin-dependent kinase (CDK) inhibitor. It inhibits the activity of cell-cycle CDKs CDK1/cyclin B (IC50 = 7 nmol/L), CDK2/cyclin E (IC50 = 9 nmol/L), CDK4/cyclin D1 (IC50 = 11 nmol/L), and transcriptional CDKs CDK9/cyclin T1 (IC50 = 5 nmol/L) and CDK7/cyclin H/MAT1 (IC50 = 25 nmol/L). It shows no off-target inhibition of carbonic anhydrase II (CA-II, IC50 > 10,000 nmol/L) [1, 2]. |
| ln Vitro |
Roniciclib (BAY-1000394; BAY1000394) inhibits the kinase activity of the cell-cycle CDKs CDK1/cyclin B, CDK2/cyclin E, and CDK4/cyclinDwith IC50 values of 7, 9, and 11 nM, respectively. The range of inhibition for the transcriptional CDKs CDK9/cyclin T1 and CDK7/cyclin H/MAT1 is comparable (5 and 25 nM)[1]. With a very balanced profile (mean IC50 on human tumor cells: 16 nM), riciclib potently inhibits the proliferation of several human and murine tumor cell lines[2]. Roniciclib inhibits proliferation of a broad spectrum of human cancer cell lines with IC50 values between 6 and 84 nmol/L (mean 16 nmol/L on a panel of 25 human cancer cell lines). On a panel of 40 human lung tumor cell lines, IC50 values range from 9 to 79 nmol/L (mean 39 nmol/L). On a panel of 24 human breast tumor and immortalized cell lines, IC50 values range from 6 to 84 nmol/L (mean 37 nmol/L). No cell line with IC50 >100 nmol/L was identified [1]. In HeLa cells, Roniciclib induces concentration-dependent increases in caspase-3/7 activity and DNA fragmentation, indicating apoptotic cell death. In wash-out experiments, IC50 values decrease from >1000 nmol/L to 14 nmol/L with increasing exposure time, with approximately 16 hours of exposure required for maximal effect [1]. Roniciclib arrests cells at multiple CDK-dependent steps within the cell cycle. In asynchronously growing HeLa cells, treatment reduces G0-G1 phase fraction from 69% to 52% and increases sub-2N DNA content from 1% to 16% (apoptotic cells). In double-thymidine block release experiments, the compound arrests cells at G1-S boundary and at various other CDK-dependent transitions [1]. Roniciclib inhibits phosphorylation of CDK downstream targets: it completely suppresses retinoblastoma protein phosphorylation at Ser780 in MCF7 cells at 30 nmol/L (indicating CDK4/2 inhibition); reduces T199-phosphorylated nucleophosmin in nocodazole-arrested HeLa cells at 30 nmol/L and abolishes it at 100 nmol/L (indicating CDK1 inhibition); and inhibits Ser2 phosphorylation of RNA polymerase II and reduces MCL-1 protein levels in A549 cells at 100 nmol/L (indicating CDK9 inhibition) [1]. BAY 1000394 exhibited broad antiproliferative activity against a panel of 25 human cancer cell lines, with IC₅₀ values ranging from 8 to 33 nM (mean 16 nM). Similar potency was observed across panels of 40 human lung tumor cell lines (mean IC₅₀ 39 nM) and 24 human breast tumor cell lines (mean IC₅₀ 37 nM), indicating uniform sensitivity regardless of genetic background (e.g., p53, pRB, K-Ras status). [1] In HeLa cells, BAY 1000394 induced apoptotic cell death in a concentration-dependent manner, as evidenced by increased caspase-3/7 activity and DNA fragmentation after 17 hours of treatment. [1] Cell cycle analysis in HeLa cells showed that BAY 1000394 (100 nM, 24h) arrested cells at various CDK-dependent steps, reducing the G0-G1 population and increasing the sub-2N (apoptotic) population. In synchronized cells released from a double-thymidine block, the compound arrested the majority of cells at the G1-S boundary. [1] Mechanistically, BAY 1000394 (30-100 nM) suppressed phosphorylation of retinoblastoma protein (pRb, a CDK2/4 substrate), reduced T199-phosphorylated nucleophosmin (a CDK1 substrate), and inhibited Ser-2 phosphorylation of RNA polymerase II (a CDK9 substrate) in various cancer cell lines. [1] |
| ln Vivo |
The T/C values at the lower dose are 0.19, indicating strong dose-dependent inhibition of tumor growth, and at the higher dose, they are 0.02 (tumor regression). Roniciclib also significantly suppresses the growth of HeLa-MaTu tumors that have grown to a size of about 50 mm2 prior to the initiation of treatment (day 8 following inoculation). Tumor growth is slowed to T/C values of 0.15 and 0.62 (respectively) by treatment with Roniciclib at 1.5 and 1 mg/kg. T/C values of 0.01 (1.0 mg/kg Roniciclib) and -0.02 (1.5 mg/kg Roniciclib) indicate a significant inhibition of tumor growth when Roniciclib is added to cisplatin[1]. Low blood clearance rates of riciclib have been reported in mice, rats, and dogs (0.51, 0.78, and 0.50 Lh-1kg-1, respectively) [2]. In athymic mice bearing HeLa-MaTu human cervical tumor xenografts, oral administration of Roniciclib once daily for 21 days produces dose-dependent tumor growth inhibition with T/C values of 0.61 at 0.5 mg/kg, 0.03 at 2 mg/kg, and signs of tumor regression at the 2 mg/kg dose. Treatment is well tolerated with no body weight loss below initial body weight [1]. On an intermittent dosing schedule (2 days on/5 days off for 3 cycles), Roniciclib at 1.5, 2.0, and 2.5 mg/kg twice daily shows dose-dependent efficacy with T/C values of 0.19, 0.02 (tumor regression), and similar results in larger tumors (~50 mm²) [1]. In the triple-negative MX-1 human breast cancer model, cisplatin-resistant A2780-cis ovarian cancer model, and multidrug-resistant OVCAR-8-ADR ovarian cancer model, Roniciclib is more efficacious than doxorubicin, cisplatin, or paclitaxel [1]. In cell line-derived and patient-derived SCLC xenograft models (NCI-H82, etc.), Roniciclib strongly inhibits tumor growth with T/C values between 0.12 and 0.19, comparable or superior to cisplatin [1]. In NCI-H82 SCLC xenografts, combination of Roniciclib (1.0-1.5 mg/kg) with cisplatin (6 mg/kg) or etoposide (12 mg/kg) produces more than additive efficacy (observed T/C values below Bliss-calculated additive values). Triple combination with cisplatin, etoposide, and Roniciclib (0.75 mg/kg) yields T/C of 0.09, significantly below expected additive T/C of 0.40, without worsening body weight loss compared to cisplatin/etoposide alone [1]. Pharmacodynamic analysis in HeLa-MaTu tumors shows that a single oral dose of Roniciclib (2 mg/kg) suppresses pRb phosphorylation for up to 7 hours, with recovery by 48 hours. Unbound drug concentrations exceed the in vitro IC50 (11 nmol/L) for approximately 24 hours [1]. |
| Enzyme Assay |
For CDK1 and CDK2 kinase assays, recombinant human CDK1/cyclin B or CDK2/cyclin E GST-fusion proteins (purified from baculovirus-infected Sf9 insect cells) are incubated with test compounds for 10 minutes at 22°C in assay buffer containing Tris-HCl, MgCl2, dithiothreitol, Na3VO4, PEG, ATP, [γ-33P]ATP, and calf thymus histone type IIIS as substrate. The reaction is stopped with EDTA solution. Reaction mixtures are transferred to P30 filter sheets, washed with phosphorous acid, and subjected to scintillation counting. IC50 values are determined as the inhibitor concentration resulting in 50% substrate phosphorylation [2]. For VEGF-R2 kinase assay, GST-tagged recombinant human VEGF-R2 kinase domain purified from Sf9 cells is incubated with test compounds for 10 minutes at 22°C in Tris-HCl buffer containing MgCl2, MnCl2, dithiothreitol, PEG, ATP, [γ-33P]ATP, and poly(Glu,Tyr) substrate. Reaction processing and IC50 determination are as described above [2]. Carbonic anhydrase II inhibition is determined using the 4-nitrophenyl acetate hydrolysis technique in a 96-well spectrophotometer [2]. |
| Cell Assay |
For proliferation assays, cells are seeded in 96-well plates at 1,000-5,000 cells/well in medium containing 10% FCS. After 24 hours, serial dilutions of Roniciclib are added and cells are incubated for 96 hours. Relative cell numbers are quantified by crystal violet staining. IC50 values are calculated using 4-parameter fit [1]. For apoptosis assays, caspase-3/7 activity is measured using ApoONE Caspase-3/7 Assay, and DNA fragmentation is measured using Cell Death Detection ELISA Plus. Results are normalized to vehicle controls and cell number [1]. For cell cycle analysis, HeLa cells are grown asynchronously or released from double-thymidine block, treated with vehicle or Roniciclib (100 nmol/L), harvested at indicated times, stained with propidium iodide, and analyzed by flow cytometry [1]. For downstream target analysis, MCF7 cells are stimulated to enter cell cycle from quiescence and treated with Roniciclib; pRb phosphorylation is analyzed by immunoblotting. Nocodazole-arrested HeLa cells are treated with Roniciclib and analyzed for phospho-nucleophosmin. A549 cells are treated with Roniciclib and analyzed for phospho-RNA polymerase II and MCL-1 levels by immunoblotting [1]. |
| Animal Protocol |
Mice: Athymic mice with established HeLa-MaTu xenograft tumors measuring approximately 25 mm2 are given oral doses of Roniciclib (BAY 1000394) once a day for 21 days. There is no weight loss below the starting body weight, indicating that the treatment is well tolerated. A cyclic intermittent dosing schedule is used to treat additional mouse groups. Doses of 1.5, 2.0, and 2.5 mg/kg are administered twice daily for two days, after which the mice receive no treatment for five days (2 on/5 off). Three treatment cycles have been finished in total[1].
For efficacy studies, female athymic nu/nu mice (50 days old, 20-22 g) are subcutaneously implanted with 1.5×10⁶ tumor cells (HeLa-MaTu, MX-1, A2780-cis, OVCAR-8-ADR, NCI-H82, or patient-derived SCLC models) suspended in 50% Matrigel/50% culture medium. When tumors reach approximately 21-25 mm², mice are randomized into treatment and control groups (8 mice/group). Roniciclib is dissolved in vehicle (40% polyethylene glycol, 60% water) and administered orally (p.o.) at indicated doses and schedules (once daily or intermittent 2 days on/5 days off). Tumor area (length × perpendicular) and body weight are measured twice weekly. Statistical analysis uses one-way ANOVA with pairwise comparison versus control [1]. For pharmacodynamic studies, mice bearing HeLa-MaTu tumors receive a single oral dose of Roniciclib (2 mg/kg). Tumors are harvested at 1, 7, 24, and 48 hours post-dose, homogenized, and analyzed for pRb phosphorylation using Meso Scale Discovery Phospho (Ser780)/Total Rb Whole Cell Lysate Kit. Unbound drug concentrations in serum are determined [1]. For combination studies, cisplatin (6 mg/kg i.p. on day 1) and/or etoposide (12 mg/kg i.p. on days 1-3 or 3-5) are administered in 14-day cycles with Roniciclib (p.o. on days 1-3, 3-5, or 8-10) at indicated doses [1]. |
| ADME/Pharmacokinetics |
Roniciclib has high aqueous solubility: thermodynamic solubility in water at pH 7.4 is 182 mg/L (22-fold higher than ZK 304709) by equilibrium shake-flask method; solubility is even higher at low pH and in organic solvents. Melting point is 134°C [2]. In vitro metabolic stability: in liver microsome preparations from human, mouse, and rat, recovery rates of parent compound after 60 min incubation are 94%, 100%, and 100%, respectively [1, 2]. Cytochrome P450 inhibition: IC50 values >20 μmol/L for all tested CYP isoenzymes [1]. Caco-2 permeability: apparent permeability coefficient (Papp ap-bas) of 79 nm/s, suggesting good intestinal permeability [1]. Plasma protein binding: 93-95% in plasma from mouse, rat, dog, and human [1]. In vivo pharmacokinetics: After intravenous administration, clearance is low across species (mouse: 0.51 L·h⁻¹·kg⁻¹; rat: 0.78 L·h⁻¹·kg⁻¹; dog: 0.50 L·h⁻¹·kg⁻¹). Volume of distribution is high (1.8-2.5 L/kg). Half-life ranges from 2.1 h (rat) to 3.7 h (mouse) to 4.3 h (dog). After oral administration, bioavailability is moderate across species (48-56%) [1, 2]. |
| Toxicity/Toxicokinetics |
In efficacy studies, Roniciclib is well tolerated at therapeutic doses (up to 2.5 mg/kg) with no significant body weight loss observed in any treatment group (once daily or intermittent schedules). In combination studies with cisplatin and etoposide, addition of Roniciclib does not worsen body weight loss compared to cisplatin/etoposide alone [1]. The compound shows no off-target inhibition of carbonic anhydrase II (IC50 >10,000 nmol/L), eliminating the erythrocyte accumulation issue seen with previous sulfonamide analogs [2]. |
| References |
[1]. BAY 1000394, a novel cyclin-dependent kinase inhibitor, with potent antitumor activity in mono- and in combination treatment upon oral application. Mol Cancer Ther. 2012 Oct;11(10):2265-73. [2]. The lab oddity prevails: discovery of pan-CDK inhibitor (R)-S-cyclopropyl-S-(4-{[4-{[(1R,2R)-2-hydroxy-1-methylpropyl]oxy}-5-(trifluoromethyl)pyrimidin-2-yl]amino}phenyl)sulfoximide (BAY 1000394) for the treatment of cancer. ChemMedChem. 2013 Jul;8(7):1067-85. |
| Additional Infomation |
Roniciclib has been investigated for the treatment of Small Cell Lung Carcinoma. Roniciclib is an orally bioavailable cyclin dependent kinase (CDK) inhibitor with potential antineoplastic activity. Roniciclib selectively binds to and inhibits the activity of CDK1/Cyclin B, CDK2/Cyclin E, CDK4/Cyclin D1, and CDK9/Cyclin T1, serine/threonine kinases that play key roles in the regulation of the cell cycle progression and cellular proliferation. Inhibition of these kinases leads to cell cycle arrest during the G1/S transition, thereby leading to an induction of apoptosis, and inhibition of tumor cell proliferation. CDKs are often dysregulated in cancerous cells. See also: Roniciclib (annotation moved to). Human Tumor Xenograft Efficacy Study: Female athymic nude mice bearing established subcutaneous HeLa-MaTu cervical cancer xenografts (tumor size ~21-25 mm²) were randomized into groups (n=8). BAY 1000394 was administered orally (per os) once daily (qd) at doses of 0.5, 1.0, 1.5, and 2.0 mg/kg for 21 consecutive days. The compound was formulated in a vehicle of 40% polyethylene glycol and 60% water. Tumor growth was monitored by caliper measurement twice a week. [1] Intermittent Dosing Study: In the same model, BAY 1000394 was administered orally twice daily (bid) at doses of 1.5, 2.0, and 2.5 mg/kg for 2 days followed by 5 days off (2 on/5 off schedule), for a total of 3 cycles. [1] Pharmacokinetic/Pharmacodynamic (PK/PD) Study: Mice bearing HeLa-MaTu tumors received a single oral dose of BAY 1000394 at 2 mg/kg (formulated as above). Tumor and plasma samples were collected at 1, 7, 24, and 48 hours post-dose for analysis of unbound drug concentration and phosphorylation levels of retinoblastoma protein (P-pRb) in tumor tissue. [1] Roniciclib (BAY 1000394) is a novel oral pan-CDK inhibitor developed to overcome the limitations of the earlier clinical candidate ZK 304709, which failed in phase I due to dose-limited absorption and high inter-patient variability attributed to low solubility and off-target carbonic anhydrase inhibition. The introduction of a sulfoximine group in Roniciclib maintains potent CDK inhibition while eliminating CA inhibitory activity and improving solubility. The compound inhibits both cell-cycle CDKs (1, 2, 4) and transcriptional CDKs (7, 9), leading to cell cycle arrest at multiple checkpoints and induction of apoptosis. It shows broad antitumor activity in various xenograft models, including chemotherapy-resistant and patient-derived models, and demonstrates more than additive efficacy when combined with cisplatin and etoposide in SCLC models. Roniciclib is efficacious on both continuous once-daily and intermittent dosing schedules, providing flexibility for clinical optimization. It is currently in phase I clinical trials for advanced solid tumors (ClinicalTrials.gov identifier: NCT01188252) [1, 2]. |
Solubility Data
| Solubility (In Vitro) | DMSO: ≥ 250 mg/mL (~580.8 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (4.83 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (4.83 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (4.83 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3232 mL | 11.6158 mL | 23.2315 mL | |
| 5 mM | 0.4646 mL | 2.3232 mL | 4.6463 mL | |
| 10 mM | 0.2323 mL | 1.1616 mL | 2.3232 mL |