Physicochemical Properties
| Molecular Formula | C29H28N6O2S |
| Molecular Weight | 524.6366 |
| Exact Mass | 524.2 |
| Elemental Analysis | C, 66.39; H, 5.38; N, 16.02; O, 6.10; S, 6.11 |
| CAS # | 1639357-93-9 |
| PubChem CID | 155908174 |
| Appearance | White to off-white solid powder |
| LogP | 3.6 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 8 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 38 |
| Complexity | 976 |
| Defined Atom Stereocenter Count | 2 |
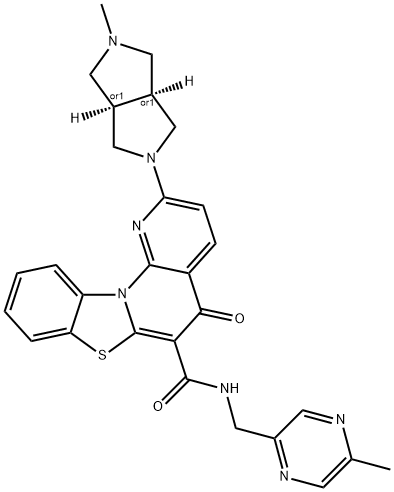
| SMILES | CC1=CN=C(C=N1)CNC(=O)C2=C3N(C4=CC=CC=C4S3)C5=C(C2=O)C=CC(=N5)C6C[C@@H]7CN(C[C@@H]7C6)C |
| InChi Key | ZAOCPDXRBFOCQL-YQQQUEKLSA-N |
| InChi Code | InChI=1S/C29H28N6O2S/c1-16-11-31-20(12-30-16)13-32-28(37)25-26(36)21-7-8-22(17-9-18-14-34(2)15-19(18)10-17)33-27(21)35-23-5-3-4-6-24(23)38-29(25)35/h3-8,11-12,17-19H,9-10,13-15H2,1-2H3,(H,32,37)/t17?,18-,19+ |
| Chemical Name | 2-[(3aR,6aS)-2-methyl-3,3a,4,5,6,6a-hexahydro-1H-cyclopenta[c]pyrrol-5-yl]-N-[(5-methylpyrazin-2-yl)methyl]-5-oxo-[1,3]benzothiazolo[3,2-a][1,8]naphthyridine-6-carboxamide |
| Synonyms | ZYN57939; ZYN-57939; ZYN 57939; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
MTR-106 targets G-quadruplex (G4) structures, with a DC₅₀ value of 0.32 μM (concentration required to stabilize 50% of G4) in FRET melting assay; it shows 25-fold selectivity for G4 over double-stranded DNA (dsDNA) [1] |
| ln Vitro |
MTR-106 (0-100 μM; 7 d) inserts into cells exhibiting anti-tumor activity in both PARPi and HR-deficient cells [1]. MTR-106 (0-100 μM; 7 d) Induction of cell cycle and work MTR-106 exhibited potent antiproliferative activity against BRCA-deficient cancer cell lines: IC₅₀ = 0.18 μM (MDA-MB-436, BRCA1⁻/⁻), IC₅₀ = 0.22 μM (HCC1937, BRCA1⁻/⁻), IC₅₀ = 0.25 μM (Capan-1, BRCA2⁻/⁻) [1] - The compound showed low cytotoxicity to BRCA-proficient cells: IC₅₀ > 10 μM (MCF-7, BRCA1⁺), IC₅₀ > 10 μM (HT-29, BRCA2⁺) [1] - MTR-106 (0.5 μM) stabilized rDNA G4 structures in cancer cells, inhibiting RNA polymerase I-mediated rRNA transcription by ~70% as detected by qRT-PCR [1] - Treatment with MTR-106 (0.2–1 μM) induced DNA damage in BRCA-deficient cells, as evidenced by increased γ-H2AX foci formation and PARP cleavage (Western blot) [1] - MTR-106 (0.5 μM) arrested BRCA-deficient cells in G2/M phase (flow cytometry) and promoted apoptosis, with apoptotic rate increased by ~35% compared to control [1] |
| ln Vivo |
BRCA-deficient and PARPi xenograft tumor growth is inhibited in nude mice by MTR-106 (10–30 mg/kg; lateral; twice weekly for 29 days) [1]. In MDA-MB-436 (BRCA1⁻/⁻) xenograft mice model, intraperitoneal injection of MTR-106 (10 mg/kg, twice weekly for 3 weeks) inhibited tumor growth by 68% compared to vehicle control [1] - MTR-106 (10 mg/kg) treatment did not cause significant weight loss (≤5% body weight change) or obvious organ damage in mice [1] - Immunohistochemical staining of tumor tissues showed MTR-106 reduced Ki-67 (proliferation marker) expression by ~50% and increased γ-H2AX expression, confirming in vivo DNA damage and anti-proliferative effects [1] |
| Enzyme Assay |
FRET melting assay: Fluorescently labeled G4 DNA or dsDNA was incubated with serial dilutions of MTR-106. The melting temperature (Tm) of DNA was measured by fluorescence intensity changes, and DC₅₀ was calculated to evaluate G4 stabilization activity [1] - RNA polymerase I transcription assay: Nuclear extracts from BRCA-deficient cells were prepared and incubated with MTR-106 and rDNA promoter template. Transcription products were quantified by qRT-PCR to assess inhibition efficiency [1] - Circular dichroism (CD) spectroscopy: G4 DNA was incubated with MTR-106, and CD spectra were recorded to confirm G4 structure stabilization and binding specificity [1] |
| Cell Assay |
cell viability assay [1] Cell Types: HR-deficient and PARPi-resistant cancer cells Tested Concentrations: 0-100 μM Incubation Duration: 7 days Experimental Results: Inhibition of HR Viability of defective cells and PARPi-. Resistant cells were dose-dependent. Cell cycle analysis [1] Cell Types: Capan-1 cells Tested Concentrations: 0.1, 0.3 and 1 μM Incubation Duration: 24 hrs (hours) Experimental Results: The number of G2/M cells increased and the number of G1 cells diminished. Western Blot Analysis[1] Cell Types: Capan-1 Cell Tested Concentrations: 1, 5 and 10 μM Incubation Duration: 24 hrs (hours) Experimental Results: Dose-dependent increase in cleaved caspases 3, 7 and 9 and cleaved PARP. Cell proliferation assay: BRCA-deficient/proficient cancer cells were seeded in 96-well plates and treated with serial dilutions of MTR-106 for 72 hours. Cell viability was measured by colorimetric assay, and IC₅₀ values were calculated [1] - Cell cycle and apoptosis assay: Cells were treated with MTR-106 (0.2–1 μM) for 48 hours, stained with propidium iodide (PI) for cell cycle analysis or Annexin V-FITC/PI for apoptosis detection, and analyzed by flow cytometry [1] - Western blot analysis: Cells treated with MTR-106 were lysed, proteins separated by SDS-PAGE, transferred to membranes, and probed with antibodies against γ-H2AX, PARP, cleaved caspase-3, and GAPDH (loading control). Band intensity was quantified by densitometry [1] - Immunofluorescence assay: Cells were treated with MTR-106, fixed, permeabilized, and incubated with anti-γ-H2AX antibody. Fluorescent foci were visualized by confocal microscopy to quantify DNA damage [1] |
| Animal Protocol |
Animal/Disease Models: BRCA-deficient and PARPi-resistant nude mouse xenografts [1] Doses: 10, 20, 30 mg/kg Route of Administration: Oral; twice weekly for 29 days Experimental Results: Tumor inhibition in a dose-dependent manner grow. Xenograft model establishment: Female nude mice (6–8 weeks old) were subcutaneously injected with MDA-MB-436 cells (5×10⁶ cells/mouse) into the right flank. Tumors were allowed to grow to ~100 mm³ before treatment [1] - Drug formulation: MTR-106 was dissolved in dimethyl sulfoxide (DMSO) and further diluted with polyethylene glycol 400 (PEG400) and normal saline (v/v/v = 1:4:5) to the desired concentration [1] - Administration protocol: Mice were randomly divided into vehicle control and drug-treated groups (n=6 per group). MTR-106 was administered via intraperitoneal injection at 10 mg/kg, twice weekly for 3 weeks [1] - Sample collection: At the end of treatment, mice were euthanized. Tumors were excised, weighed, and fixed in formalin for immunohistochemical analysis; major organs (heart, liver, spleen, lung, kidney) were collected for pathological examination [1] |
| Toxicity/Toxicokinetics |
In vitro toxicity: MTR-106 showed low cytotoxicity to normal human fibroblasts (IC₅₀ > 10 μM) [1] - In vivo toxicity: No significant abnormalities were observed in pathological sections of major organs from MTR-106-treated mice; plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were within normal ranges [1] - Plasma protein binding rate: MTR-106 bound to plasma proteins at a rate of ~89% as determined by ultrafiltration method [1] |
| References |
[1]. Discovery of MTR-106 as a highly potent G-quadruplex stabilizer for treating BRCA-deficient cancers. Invest New Drugs. 2021 Oct;39(5):1213-1221. |
| Additional Infomation |
MTR-106 is a synthetic small-molecule compound designed as a highly selective G-quadruplex stabilizer [1] - Its anticancer mechanism involves stabilizing rDNA G-quadruplexes, inhibiting RNA polymerase I transcription, inducing nucleolar stress and DNA damage, and ultimately leading to cell cycle arrest and apoptosis in BRCA-deficient cancers [1] - MTR-106 exhibits synthetic lethality with BRCA deficiency, making it a potential targeted therapy for BRCA-mutated breast, ovarian, and pancreatic cancers [1] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9061 mL | 9.5303 mL | 19.0607 mL | |
| 5 mM | 0.3812 mL | 1.9061 mL | 3.8121 mL | |
| 10 mM | 0.1906 mL | 0.9530 mL | 1.9061 mL |