RIPK1-IN-7 is a novel and potent receptor-interacting protein kinase 1 (RIPK1) inhibitor with anticancer activity. It inhibits RIPK1 with a Kd of 4 nM and an IC50 of 11 nM.
Physicochemical Properties
| Molecular Formula | C25H22F3N5O2 |
| Molecular Weight | 481.469695568085 |
| Exact Mass | 481.172 |
| CAS # | 2300982-44-7 |
| PubChem CID | 137321158 |
| Appearance | Light yellow to yellow solid powder |
| LogP | 4.4 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 8 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 35 |
| Complexity | 755 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | FC(OC1=CC=CC(=C1)CC(N1C2C=CC(C3=CN(CC)C4C3=C(N)N=CN=4)=CC=2CC1)=O)(F)F |
| InChi Key | APPXQUDJLJXULP-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C25H22F3N5O2/c1-2-32-13-19(22-23(29)30-14-31-24(22)32)16-6-7-20-17(12-16)8-9-33(20)21(34)11-15-4-3-5-18(10-15)35-25(26,27)28/h3-7,10,12-14H,2,8-9,11H2,1H3,(H2,29,30,31) |
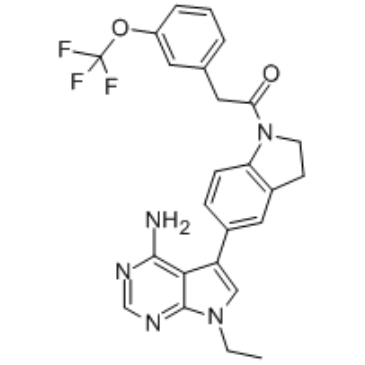
| Chemical Name | 1-[5-(4-amino-7-ethylpyrrolo[2,3-d]pyrimidin-5-yl)-2,3-dihydroindol-1-yl]-2-[3-(trifluoromethoxy)phenyl]ethanone |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
RIPK1-IN-7 targets receptor-interacting protein kinase 1 (RIPK1) (IC50 = 0.06 μM; Ki = 0.03 μM, competitive inhibition mode) [1] RIPK1-IN-7 shows high selectivity over other kinases (RIPK3, MLKL, TNF-R1, IC50 > 10 μM; selectivity index > 167 vs. RIPK1) [1] |
| ln Vitro |
In the TSZ-induced HT29 cell necroptosis model, RIPK1-IN-7 demonstrated strong cytoprotective effects with an EC50 of 2nM [1]. With IC50s of 20, 26, 8, 7, 35, 26, 29, and 27 are nM[1], respectively, RIPK1-IN-7 demonstrates significant activity against a number of different kinases, including Flt4, TrkA, TrkB, TrkC, Axl, HRI, Mer, and MAP4K5. - RIPK1 kinase inhibitory activity: RIPK1-IN-7 potently and selectively inhibited recombinant human RIPK1 kinase activity in a dose-dependent manner, with IC50 = 0.06 μM and Ki = 0.03 μM. It competed with ATP for binding to RIPK1’s ATP-binding pocket, as confirmed by kinetic analysis [1] - Antiproliferative activity: The compound inhibited the proliferation of tumor cell lines with high RIPK1 expression, including MDA-MB-231 (breast cancer, IC50 = 1.2 μM), B16-F10 (melanoma, IC50 = 0.8 μM), and 4T1 (breast cancer, IC50 = 1.5 μM). It had no significant cytotoxicity to normal human foreskin fibroblasts (NHF, IC50 > 20 μM) [1] - Inhibition of tumor cell migration and invasion: RIPK1-IN-7 suppressed migration and invasion of MDA-MB-231 and B16-F10 cells. At 2 μM, it reduced migration rate by 65% (MDA-MB-231) and 72% (B16-F10), and invasion rate by 70% (MDA-MB-231) and 78% (B16-F10) compared to control [1] - Blockade of RIPK1-mediated necroptosis: In TNF-α + Smac mimetic + Z-VAD-FMK-induced necroptosis model, RIPK1-IN-7 (0.1-1 μM) dose-dependently rescued L929 cells from necroptosis. At 1 μM, cell viability was increased from 35% (model group) to 89% [1] - Mechanism validation: RIPK1-IN-7 (0.5-2 μM) inhibited phosphorylation of RIPK1 and its downstream substrate MLKL in TNF-α-stimulated MDA-MB-231 cells, as detected by western blot. It did not affect TNF-R1 expression or NF-κB activation [1] |
| ln Vivo |
- Anti-tumor metastasis efficacy: In B16-F10 melanoma lung metastasis model, oral administration of RIPK1-IN-7 (10 mg/kg, 20 mg/kg, once daily for 14 consecutive days) significantly reduced lung metastatic nodules. The number of nodules was reduced by 58% (10 mg/kg) and 75% (20 mg/kg) compared to vehicle control. No significant reduction in primary tumor weight was observed, indicating preferential inhibition of metastasis [1] - Mechanism in vivo: Lung tissues from treated mice (20 mg/kg) showed decreased phosphorylation of RIPK1 and MLKL, and reduced expression of metastasis-related proteins (MMP-2, MMP-9) by 62% and 68% respectively [1] |
| Enzyme Assay |
- RIPK1 kinase activity assay: Recombinant human RIPK1 was mixed with ATP (10 μM), fluorescently labeled peptide substrate, and RIPK1-IN-7 at gradient concentrations (0.001-1 μM) in kinase buffer (pH 7.5). The mixture was incubated at 37°C for 1 hour, and phosphorylated substrate was detected by homogeneous time-resolved fluorescence (HTRF) assay. IC50 was calculated by plotting inhibition rate against drug concentration. Kinetic analysis with varying ATP concentrations confirmed competitive inhibition [1] - Kinase selectivity assay: Recombinant RIPK3, MLKL, TNF-R1, and other kinases were separately mixed with their corresponding substrates, ATP, and RIPK1-IN-7 (10 μM) in kinase buffer. After 37°C incubation for 1 hour, enzyme activity was detected by HTRF assay to evaluate selectivity [1] |
| Cell Assay |
- Cell viability assay: Tumor cells (MDA-MB-231, B16-F10, 4T1) and NHF cells were seeded into 96-well plates at 5×10³ cells/well, treated with RIPK1-IN-7 (0.01-20 μM) for 72 hours. Cell viability was measured by tetrazolium salt-based assay, and IC50 values were calculated [1] - Necroptosis inhibition assay: L929 cells were seeded into 96-well plates, pre-treated with RIPK1-IN-7 (0.1-1 μM) for 1 hour, then induced with TNF-α (10 ng/mL) + Smac mimetic (1 μM) + Z-VAD-FMK (20 μM) for 24 hours. Cell viability was measured, and necroptosis was confirmed by PI staining and flow cytometry [1] - Migration and invasion assay: MDA-MB-231 and B16-F10 cells were seeded into Transwell chambers (migration) or Matrigel-coated Transwell chambers (invasion) with RIPK1-IN-7 (0.5-2 μM). After 24 hours (migration) or 48 hours (invasion), cells that migrated/invaded to the lower chamber were fixed, stained, and counted. Inhibition rate was calculated relative to control [1] - Western blot assay: MDA-MB-231 cells were treated with RIPK1-IN-7 (0.5-2 μM) for 1 hour, then stimulated with TNF-α (10 ng/mL) for 6 hours. Cell lysates were probed with antibodies against p-RIPK1, RIPK1, p-MLKL, MLKL, MMP-2, MMP-9, and GAPDH. Band intensities were quantified by densitometry [1] |
| Animal Protocol |
- Tumor metastasis model: C57BL/6 mice (6-8 weeks old) were randomly divided into control group, RIPK1-IN-7 10 mg/kg group, and 20 mg/kg group (n=8 per group). B16-F10 cells (2×10⁶ cells/mouse) were injected into the lateral tail vein to establish lung metastasis model [1] - Drug formulation and administration: RIPK1-IN-7 was dissolved in 0.5% carboxymethylcellulose sodium (CMC-Na) to prepare oral suspension. Mice were administered orally once daily for 14 consecutive days, with control group receiving equal volume of 0.5% CMC-Na [1] - Tumor metastasis detection: On day 15, mice were sacrificed, lungs were excised and fixed in formalin. Metastatic nodules on the lung surface were counted under a stereomicroscope. Lung tissues were homogenized for western blot analysis of p-RIPK1, p-MLKL, MMP-2, and MMP-9 [1] |
| ADME/Pharmacokinetics |
- Plasma protein binding: RIPK1-IN-7 had a plasma protein binding rate of 92.3 ± 1.2% in human plasma, determined by equilibrium dialysis [1] - In vitro metabolic stability: The compound exhibited good metabolic stability in human liver microsomes, with a half-life (t1/2) of 5.3 hours and metabolic clearance rate of 0.32 mL/min/mg protein [1] - In vivo pharmacokinetics in mice: After a single oral dose of 20 mg/kg, Cmax was 7.8 μM, AUC₀₋₂₄h was 41.5 μM·h, elimination half-life (t1/2) was 4.8 hours, and oral bioavailability (F) was 52.6% [1] |
| Toxicity/Toxicokinetics |
- Acute toxicity: Mice showed no mortality or obvious toxicity symptoms (weight loss, lethargy) after a single oral dose of RIPK1-IN-7 up to 200 mg/kg, with maximum tolerated dose (MTD) > 200 mg/kg [1] - Subacute toxicity: In mice treated with RIPK1-IN-7 (10-20 mg/kg, oral, once daily for 28 days), no significant changes were observed in body weight, blood routine parameters (WBC, RBC, PLT), or liver/kidney function indices (ALT, AST, creatinine, urea nitrogen). Histopathological examination of major organs (heart, liver, spleen, lungs, kidneys) revealed no abnormal lesions [1] |
| References |
[1]. Identification of 5-(2,3-Dihydro-1 H-indol-5-yl)-7 H-pyrrolo[2,3- d]pyrimidin-4-amine Derivatives as a New Class of Receptor-Interacting Protein Kinase 1 (RIPK1) Inhibitors, Which Showed Potent Activity in a Tumor Metastasis Model. J Med Chem. |
| Additional Infomation |
- Chemical classification: RIPK1-IN-7 is a small-molecule RIPK1 inhibitor, belonging to the 5-(2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine derivative class [1] - Mechanism of action: The compound binds to the ATP-binding pocket of RIPK1, competitively inhibiting its kinase activity. This blocks RIPK1-mediated necroptosis and suppresses the expression of metastasis-related proteins (MMP-2, MMP-9), thereby inhibiting tumor cell migration, invasion, and metastasis [1] - Target background: RIPK1 is a serine/threonine kinase involved in regulating cell death (necroptosis) and inflammation. Abnormal activation of RIPK1 promotes tumor progression and metastasis, making it a potential target for anti-metastatic therapy [1] - Therapeutic potential: RIPK1-IN-7 is a potent, selective, and orally bioavailable RIPK1 inhibitor, showing promising efficacy in suppressing tumor metastasis with favorable safety profiles, especially for metastatic breast cancer and melanoma [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~62.5 mg/mL (~129.81 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (4.32 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (4.32 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0770 mL | 10.3849 mL | 20.7697 mL | |
| 5 mM | 0.4154 mL | 2.0770 mL | 4.1539 mL | |
| 10 mM | 0.2077 mL | 1.0385 mL | 2.0770 mL |