Physicochemical Properties
| Molecular Formula | C8H16NO5P |
| Molecular Weight | 237.19 |
| Exact Mass | 237.077 |
| Elemental Analysis | C, 40.51; H, 6.80; N, 5.91; O, 33.73; P, 13.06 |
| CAS # | 127910-32-1 |
| PubChem CID | 6372334 |
| Appearance | Typically exists as solid at room temperature |
| Density | 1.312 g/cm3 |
| Boiling Point | 434.1ºC at 760 mmHg |
| Melting Point | 236-239ºC |
| Flash Point | 216.3ºC |
| Vapour Pressure | 9.46E-09mmHg at 25°C |
| Index of Refraction | 1.512 |
| LogP | 0.701 |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 15 |
| Complexity | 295 |
| Defined Atom Stereocenter Count | 0 |
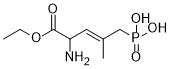
| SMILES | CCOC(C(/C=C(/CP(=O)(O)O)\C)N)=O |
| InChi Key | OKDOWCKDTWNRCB-GQCTYLIASA-N |
| InChi Code | InChI=1S/C8H16NO5P/c1-3-14-8(10)7(9)4-6(2)5-15(11,12)13/h4,7H,3,5,9H2,1-2H3,(H2,11,12,13)/b6-4+ |
| Chemical Name | [(E)-4-amino-5-ethoxy-2-methyl-5-oxopent-2-enyl]phosphonic acid |
| Synonyms | Cgp-39551; Cgp39551; DTXSID401211432; 1-Ethyl (3E)-2-amino-4-methyl-5-phosphono-3-pentenoate; DTXCID001642794; Cgp 39,551; 127910-32-1; (E)-(4-Amino-5-ethoxy-2-methyl-5-oxopent-2-en-1-yl)phosphonic acid; [(E)-4-amino-5-ethoxy-2-methyl-5-oxopent-2-enyl]phosphonic acid; 2-Amino-4-methyl-5-phosphono-3 pentenoic acid carboxyethyl ester; Cgp 39551 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | NMDA/N-methyl-D-aspartate receptor |
| ln Vitro |
With a Ki of 310 nM, CGP 39551 prevents the selective NMDA receptor antagonist [3H]-CPP from binding to postsynaptic densities (PSDs) in rat brain[2]. Receptor binding profiles of CGP 37849 and CGP 39551 [2] The high affinity of CGP 37849 for the NMDA receptor was identified on the basis of its activity at NMDA-sensitive L-[3H]-glutamate binding sites in crude PSDs (Foster & Fagg, 1987a; Fagg & Baud, 1988) and subsequently was confirmed using the selective radioligand, [3H]-CPP (Olverman et al., 1986; Murphy et al., 1987). Systematic analyses of a series of co-phosphono amino acid analogues revealed that CGP 37849 was a highly potent inhibitor of L-[3H]-glutamate binding, with a K, similar to that of Lglutamate itself Inhibitory activity was found to be associated with the trans configuration of the APPA molecule (not with the naturally-occurring cis-isomer) and, as in the case of AP5 (Table 1; Evans et al., 1982) and other NMDA receptor antagonists (Watkins et al., 1990), with the D-stereoisomer (CGP 40116). Saturation analyses indicated that the interaction of CGP 37849 with the L-[3H]-glutamate binding site was of a competitive nature; increasing concentrations of the blocker did not alter the maximum number of binding sites labelled by the radioligand, but progressively decreased the apparent affinity of L-[3H]-glutamate for the receptor (Figure 2). CGP 37849 inhibited the binding of the selective NMDA receptor antagonist, [3H]-CPP, to rat brain PSDs with a Ki of 35 nM (Figure 3 and Table 1), and was thus some 4, 5 and 7 fold more potent than the antagonists CGS 19755, CPP and D-AP5, respectively. As observed when using L-[3H]-glutamate as radioligand, activity was found to reside essentially exclusively in the D-isomer (CGP 40116, Ki 19 nM; Table 1), which showed an affinity roughly 150 fold greater than that of the L-enantiomer (CGP 40117). The ethylester, CGP 39551, showed measurable inhibitory activity at both L-[3H]-glutamate (K; 8.4 pM) and [3H]-CPP binding sites (Ki 0.31 pM), but was weaker than the free acid, CGP 37849 (Table 1). Both CGP 37849 and CGP 39551 were weak or inactive in receptor binding assays for 18 other neurotransmitters or modulators. At a concentration of 50 pM, CGP 37849 inhibited strychnine-insensitive [3H]-glycine binding by 26 + 2% and CGP 39551 by 22 + 2% (means + s.e.mean, n = 3). In the remaining 17 receptor assays used (see Methods), both compounds showed 20% inhibition of binding or less at the test concentration of 10pM. CGP 37849 thus is minimally 300 fold more selective for the NMDA receptor recognition site than for any of the other sites examined [2]. Neuropharmacological actions of CGP 37849 and CGP 39551 in vitro [2] Agonist-antagonist interaction Electrophysiological studies using the hippocampal slice preparation indicated that CGP 37849 showed no excitatory activity, but at concentrations as low as 0.3 JM antagonized the increases in CA1 pyramidal cell firing rate induced by sub-maximal concentrations of NMDA (Figure 4a). This effect was rapid in onset and in offset, and was agonist-selective, in that responses evoked by quisqualate and kainate were unaffected. Thus, the potency and selectivity of CGP 37849 as an antagonist of NMDA receptor responses is in good agreement with its mechanism of action as determined from radioligand binding studies. Effects of CGP 37849 and CGP 39551 on the uptake and release ofL-glutamate [2] Alterations of synaptic function elicited by CGP 37849 and CGP 39551 (see below) potentially might result not simply from their interaction with postsynaptic NMDA receptors, but also from modifications of glutamate uptake or release. In experiments to evaluate these possibilities, no effects of the compounds were observed. As shown by other investigators (Davies & Johnston, 1976; Johnston et al., 1979), L-[3H]-glutamate was accumulated with high affinity (Km 11 pIM) by rat brain synaptosomes and its uptake was inhibited by the reference compounds, D-aspartate and dihydrokainate (IC50 s 7 pM and 315 pM, respectively; mean values, 2 separate experiments). However, neither CGP 37849 nor CGP 39551, at concentrations of 1-10001pm, impaired L-[3H]-glutamate uptake. For example, in the presence of 10 pM CGP 37849 or CGP 39551, L-[3H]-glutamate uptake was respectively 104% and 100% of control values, and in the presence of 1 mm drug, 102% and 106%, respectively (means, 2 separate experiments, each in duplicate). Similarly, neither CGP 37849 nor CGP 39551 modified the electrically-evoked release of endogenous glutamate from rat hippocampal slices. In these experiments, basal glutamate release routinely was in the range 0.2-0.4% of the tissue content per fraction, and this was increased 2.5 + 0.2 times by 10Hz electrical field stimulation. Under control conditions, the ratio S2/S1 (release evoked by the second stimulation/ release evoked by the first stimulation) was close to unity (1.2), and this ratio was not altered by inclusion of 10pM CGP 37849 (1.1) or CGP 39551 (1.0) in the superfusion medium (mean values, 2 separate experiments, each in triplicate) [2]. Epileptiform activity [2] Stimulation of Schaffer collateralcommissural fibres in hippocampal slices bathed in low Mg2 I_ containing medium previously has been shown to evoke multiple population spikes in CAI pyramidal neurones that are reminiscent of an epileptiform burst (Coan & Collingridge, 1985; Lester et al., 1988; see Figure 4b,i). Bath application of CGP 37849 at concentrations up to 10pM markedly suppressed this type of repetitive firing, while having no effect on the first population spike of the burst (Figure 4bii). This effect was rapidly reversible, with the control pattern of bursting being restored within 10min following drug wash-out (Figure 4b,iii). CGP 39551 also suppressed stimulus-induced burst firing in this experimental paradigm, but was weaker than CGP 37849 (not shown). |
| ln Vivo |
With an ED50 of 4 mg/kg (po), CGP 39551 causes maximal electroshock-induced seizures in mice[2]. Adult rats who receive long-term CGP 39551 therapy as newborns exhibit more behavioral reactions to D2 dopamine receptor stimulation[3].
1. The pharmacological properties of CGP 37849 (DL-(E)-2-amino-4-methyl-5-phosphono-3-pentenoic acid; 4-methyl-APPA) and its carboxyethylester, CGP 39551, novel unsaturated analogues of the N-methyl-D-aspartate (NMDA) receptor antagonist, 2-amino-5-phosphonopentanoate (AP5), were evaluated in rodent brain in vitro and in vivo. 2. Radioligand binding experiments demonstrated that CGP 37849 potently (Ki 220 nM) and competitively inhibited NMDA-sensitive L-[3H]-glutamate binding to postsynaptic density (PSD) fractions from rat brain. It inhibited the binding of the selective NMDA receptor antagonist, [3H]-((+/-)-3-(2-carboxypiperazin-4-yl)propyl-1-phosphonate (CPP), with a Ki of 35 nM, and was 4, 5 and 7 fold more potent than the antagonists [+/-)-cis-4-phosphonomethylpiperidine-2-carboxylic acid) (CGS 19755), CPP and D-AP5, respectively. Inhibitory activity was associated exclusively with the trans configuration of the APPA molecule and with the D-stereoisomer. CGP 39551 showed weaker activity at NMDA receptor recognition sites and both compounds were weak or inactive at 18 other receptor binding sites. 3. CGP 37849 and CGP 39551 were inactive as inhibitors of L-[3H]-glutamate uptake into rat brain synaptosomes and had no effect on the release of endogenous glutamate from rat hippocampal slices evoked by electrical field stimulation. 4. In the hippocampal slice in vitro, CGP 37849 selectively and reversibly antagonized NMDA-evoked increases in CA1 pyramidal cell firing rate. In slices bathed in medium containing low Mg2+ levels, concentrations of CGP 37849 up to 10 microM suppressed burst firing evoked in CAl neurones by stimulation of Schaffer collateral-commissural fibres without affecting the magnitude of the initial population spike; CGP 39551 exerted the same effect but was weaker. In vivo, oral administration to rats of either CGP 37849 or CGP 39551 selectively blocked firing in hippocampal neurones induced by ionophoreticallyapplied NMDA, without affecting the responses to quisqualate or kainate. 5. CGP 37849 and CGP 39551 suppressed maximal electroshock-induced seizures in mice with ED50 s of 21 and 4 mg kg'- p.o., respectively. 6. CGP 37849 and CGP 39551 are potent and competitive NMDA receptor antagonists which show significant central effects following oral administration to animals. As such, they may find value as tools to elucidate the roles of NMDA receptors in brain function, and potentially as therapeutic agents for the treatment of neurological disorders such as epilepsy and ischaemic brain damage in man.[2] Anticonvulsant properties of CGP 37849 and CGP 39551 [2] In contrast to AP5, AP7, CPP and CGS 19755, which were ineffective anticonvulsants when given orally, CGP 37849 suppressed maximal electroshock-induced seizures in mice following oral administration with an ED50 of 21 mgkg-1 (Table 1). In agreement with radioligand binding studies at the NMDA receptor (see above), anticonvulsant activity was found to reside in the D-stereoisomer (CGP 40116; ED50 7mgkg-1). Unlike the investigations in vitro, however, the ethylester of CGP 37849 (CGP 39551; ED50 4mgkg-1) was more potent than the free acid. Details of the anticonvulsant properties of CGP 37849 and CGP 39551 will be published elsewhere. Antagonism of NMDA-evoked responses by CGP 37849 and CGP 39551 in vivo [2] Since CGP 37849 and CGP 39551 exhibited anticonvulsant activity following oral administration, it was of interest, in terms of understanding their mechanism of action in vivo, to determine whether they antagonized NMDA-evoked responses after such treatments. Figure 5 shows that this is 7. 6 5- CD a) 'EL 3.- 1n 2 a indeed the case. At doses some 5-7 times greater than their oral ED50 s as anticonvulsants, both drugs selectively blocked the responses of hippocampal neurones to ionophoreticallyapplied NMDA, without affecting responses to quisqualate or kainate (or acetylcholine, not shown). For CGP 37849, the onset of this effect was 30-40min after administration, with almost complete blockade of NMDA-evoked responses after 75-85 min. CGP 39551 showed a longer latency to onset; clear antagonism of NMDA responses was first observed 80- 90min after oral dosage, and responses were essentially abolished after 110- 120 min. Following chronic neonatal treatment with the competitive N-methyl-D-aspartate antagonist CGP 39551, adult rats showed increased behavioral responses to the D2 dopamine receptor stimulation. In nucleus accumbens and in n. striatum of similarly treated rats increases in D2 dopamine receptor number were observed. CGP 39551 was administered daily to neonatal rats with increasing doses from postnatal day 1 to 22. At postnatal days 70-82, the rats were observed for hyperactivity induced by the selective D2 dopamine receptor agonist LY 171555, the grooming behavior elicited by the specific D1 dopamine receptor stimulating agent SKF 38393 and the stereotypies induced by the mixed D1/D2 receptor agonist apomorphine. [3H]Spiroperidol and [3H]SCH 23390 specific binding to membranes of nucleus accumbens, nucleus striatum and frontal cortex of similarly treated rats was measured. The hypermotility and the stereotyped behavior induced by LY 171555 and apomorphine, respectively, were augmented, whereas grooming behavior elicited by SKF 38393 was unaffected, in CGP 39551-treated rats. Consistently, both in nucleus accumbens and in n. striatum an increase in [3H]Spiroperidol specific binding was observed, while [3H]SCH 23390 specific binding did not change. The study demonstrates that chronic blockade of N-methyl-D-aspartate receptor during the critical period of brain maturation results in long-lasting dopaminergic functional changes [3]. |
| Enzyme Assay |
NMDA receptor assays [2] Subcellular fractions enriched in postsynaptic densities (PSDs) were isolated from the brains of adult male Tif:RAIf(SPF) rats (180-250g) as previously described (Foster & Fagg, 1987a). In brief, synaptic plasma membranes were isolated by sucrose density gradient centrifugation, treated with 0.4% Triton X-100, subjected to one freeze-thaw cycle, and washed 4 times by resuspension and centrifugation in 0.5 mm HEPES-KOH buffer (pH 7.0). The resultant crude PSDs were stored as a suspension at -800C until assay, at which time they were thawed, washed once more and resuspended in assay buffer. The binding of L-[3,4-3H]-glutamic acid (44.1Cimmol-'), [1,2-3H]-CPP (20 Ci mmol- 1) (Olverman et al., 1986; Murphy et al., 1987) and [2-3H]-glycine (43.5Cimmol-t) (Bristow et al., 1986; Kessler et al., 1989) was determined essentially according to previously published procedures. Aliquots (40- 50pg protein) of crude PSDs were incubated in triplicate with radioligand (50 nM L-[3H]-glutamate; 25 nm [3H]-CPP; 20nm [3H]-glycine) in a final volume of 0.5ml 50mM Tris-acetate buffer (pH 6.9 for L-[3H]-glutamate; pH 7.8 for [3H]-CPP; pH 7.5 for [3H]-glycine), and membrane-bound radioactivity was measured after centrifugation and aspiration of the supernatant. Incubations were 30min at 32°C for L-[3H]-glutamate and [3H]-CPP, and 20 min at 4°C for [3H]-glycine. Non-specific binding was determined in the presence of 0.5 mM L-glutamate, 0.1 mM CPP or 0.5 mm glycine, and was routinely about 15%, 30% and 25% of total binding, respectively. Potential inhibitors were added to the incubation tubes prior to the addition of radioligand. IC50 values were determined by fitting data to the logistic equation using an iterative curve fitting procedure operated on a VAX 8600 computer, and Ki values were calculated from the relationship: Ki = IC54/(l + F/Kd), where F is the free concentration of radioligand and Kd its equilibrium dissociation constant (Kd values were determined by saturation analyses of the type illustrated in Figure 2; mean values used here were 250nM for L-[3H]- glutamate, and 130 nM for [3H]-CPP). Other receptors [2] Radioreceptor assays were conducted by use of procedures which have been established and documented previously (Foster et alt, 1981; Honore & Nielsen, 1985; Lehmann et al., 1988). Assays (and radioligands) used were as follows: quisqualate (AMPA), kainate (kainate), GABAA (muscimol), benzodiazepine (flunitrazepam), GABAB (CGP 27492), adenosine (cyclohexyladenosine), muscarinic cholinoceptor (diozalane and QNB), xl-adrenoceptor (prazosin), a2-adrenoceptor (clonidine), f0-adrenoceptor (dihydroalprenolol), 5-HT1 (5-HT), 5-HT2 (ketanserin), histamine H1 (doxepine), histamine H2 (thiotidine), substance P (substance P) and neurotensin (neurotensin). All assays were validated using standard reference compounds. Transport studies The Na+-dependent uptake of L-[3,4-3H]-glutamate (52.6 Ci mmol'-) into crude synaptosomal (P2) fractions isolated from rat brain was investigated by the method described by Yunger et al., (1984) for [3H]-GABA uptake with minor modifications. In brief, the P2 fraction (recovered as a pellet in 0.32M sucrose) was suspended in 0.32 M glucose and diluted with Krebs-Ringer bicarbonate buffer (composition in mM: NaCl 118, KCI 4.8, CaCl2 2.7, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, final glucose concentration 8, equilibrated with 5% CO2 in 02) containing 0.1 mM isoniazid and L-methionine sulphoximine to inhibit the metabolism of L-[3H]-glutamate. Aliquots of this P2 suspension were preincubated in duplicate (with test compounds as appropriate) at 37°C for 15min before addition of L-[3H]- glutamate (final concentration 1 pM, except for Km determinations). 'Blank' tubes were incubated on ice. Incubations were continued for a further 3 min, and were terminated by rapid filtration (0.45 pm pore size) and washing twice with ice-cold buffer. Radioactivity retained on the filter was determined by liquid scintillation counting. Transmitter release experiments [2] The electrically-evoked release of endogenous glutamate from rat hippocampal slices was determined by a slight modification of the method described by Waldmeier et al. (1988) for the release of endogenous GABA. Cross-chopped slices (about 0.36 x 0.36 x 1 mm) were suspended in a Krebs-Ringer bicarbonate buffer (composition as for Transport studies above, except that the final concentrations of CaCl2 and D-glucose were 1.8mm and 10mm, respectively) equilibrated with 5% CO2 in °2, and aliquots were transferred to superfusion chambers with platinum ring electrodes above and below the tissue slices. Slices were superfused with buffer (0.25 ml min-1, 37°C) containing 0.5mm dihydrokainate (to inhibit reuptake) for 1 h before collecting 1.25 ml fractions for amino acid analysis. The glutamate content of the superfusate and of the slices (collected at the end of each experiment) were determined by high performance liquid chromatography (h.p.l.c.) after pre-column derivatization with o-phthalaldehyde, and the glutamate released in each fraction was expressed as a percentage of the tissue content at the time of collection (% fractional release). Previous studies have shown that, in this system, the evoked release of endogenous glutamate is frequency-dependent over the range 2-40 Hz and is entirely Ca2 f-dependent (Thedinga et al., 1989). For the experiments described here, two trains of monophasic pulses (1O Hz, 20 mA, 2 ms, 2 min duration) were delivered during fractions 3 (Sj and 9 (S2); the first stimulation was always in superfusion buffer alone, and the second was either in the absence or the presence of test compounds (added to the buffer 20 min previously). Drug effects on evoked release were determined by comparing the ratio S2/ S1 in drug-treated and control slices (Waldmeier et al., 1988). All experiments were conducted in triplicate. Electrophysiological investigations [2] In vitro Experiments were performed on transverse hippocampal slices (450pm) prepared from male Tif:RAIf(SPF) rats (150-200g weight) as previously described (Olpe & Lynch, 1982). Slices were allowed to stabilize in a humidified interface chamber, and were subsequently perfused (6 ml min- ; equivalent to 2 chamber volumes per min) with a physiological salt solution (composition in mM: NaCl 124, KCl 2.5, CaCl2 2.5, MgSO4 2, KH2PO4 1.25, NaHCO3 26, D-glucose 10; gassed with 5% CO2 in 02) at 320C. Extracellular recordings were made from the stratum pyramidale region of area CAI by use of glass microelectrodes filled with 4M NaCl. Two series of experiments were performed: (1) Agonistantagonist interactions were investigated by applying submaximal concentrations (10pM) of NMDA, quisqualate or kainate at intervals of 5-10 min in the presence and absence of test blockers, and by plotting the firing rate of single neurones (action potentials in 5-10 s bins) on a chart recorder. All substances were applied via the perfusion system to submerged hippocampal slices. (2) The ability of test drugs to block synaptically-evoked epileptiform activity was evaluated using an approach employed by Coan & Collingridge (1985). Stable single population spikes were first elicited in CA1 neurones by stimulation of Schaffer collateral-commissural fibres (submerged slices), and the Mg2+ concentration of the perfusion medium was then decreased from 2 mm to nominally 0.1 mm (conditions which reduce the voltage-dependent blockade of the NMDA receptor ion channel). Subsequent sub-maximal stimulation (at a frequency of 0.125 Hz) evoked bursts of population spikes, which previously have been shown to be sensitive to blockade by NMDA receptor antagonists (Coan & Collingridge, 1985; Lester et al., 1988). |
| Animal Protocol |
In vivo [2] Male rats (Tif:RAII(SPF), 280-320g weight) were anaesthetized with chloral hydrate (400mg kg- 1, i.p.) and the tips of four-barrelled micropipettes were stereotaxically positioned in the region of the hippocampal CAI pyramidal neurones. Three barrels of the pipette contained NMDA, quisqualate and kainate (0.01 M, pH 10) and the fourth recording barrel 4 M NaCl. Excitatory amino acids were applied ionophoretically (currents 20-3OnA; adjusted to yield similar response amplitudes) for periods of 20-60s at constant intervals of 2-3min (retaining currents of 20nA were employed between drug applications). The number of action potentials in 5-lOs bins was plotted on a chart recorder, and responses to ionophoretically-applied agonists were monitored for up to 8h. Antagonists were dissolved in physiological saline and were administered cumulatively (3 h intervals) via a canula in the oesophagus. Anticonvulsant evaluation [2] Convulsions in mice were elicited by maximal electroshock (Bernasconi et al., 1986). Brief current pulses (16mA, 50 Hz, 0.2s) were applied via corneal electrodes and the number of animals displaying tonic hind limb extension seizures after saline or drug administration (5 animals per dose group) was determined. Test anticonvulsants were administered orally (via an oesophageal cannula) 2-4h before electroshock. ED50 values (dose which prevented seizures in 50% of animals) were determined by the method of Spearman-Kaerber. |
| References |
[1]. CGP 37849 and CGP 39551: novel competitive N-methyl-D-aspartate receptor antagonists with potent oral anticonvulsant activity. Prog Clin Biol Res. 1990;361:421-7. [2]. CGP 37849 and CGP 39551: novel and potent competitive N-methyl-D-aspartate receptor antagonists with oral activity. Br J Pharmacol. 1990 Apr;99(4):791-7. [3]. Chronic neonatal blockade of N-methyl-D-aspartate receptor by CGP 39551 increases dopaminergic function in adult rat. Neuroscience. 1994 Nov;63(2):451-5. |
| Additional Infomation |
CGP 37849 (or 4-methyl-APPA) is a novel competitive antagonist at the NMDA-preferring sub-type of excitatory amino acid receptor and, together with its carboxyethylester (CGP 39551), is the first agent of this pharmacological class reported to show significant central effects following oral administration in animals. The NMDA receptor-active properties of CGP 37849 were identified on the basis of its high affinity for L-[3H]-glutamate binding sites in rat brain PSDs (predominantly NMDA-sensitive sites, see Foster & Fagg, 1987a; Fagg & Baud, 1988) and were confirmed using the selective NMDA receptor radioligand, [3H]-CPP (Olverman et al., 1986; Murphy et al., 1987). In the hippocampal slice preparation, it showed no agonist activity, but antagonized NMDA-evoked increases in neuronal firing at concentrations within the range shown to inhibit radioligand binding to the NMDA receptor recognition site; in addition, its ability to suppress burst firing in CAI pyramidal neurones without affecting the fast excitatory postsynaptic potential (e.p.s.p.) or population spike is in accordance with the properties of NMDA receptor antagonists as established by other investigators (Collingridge et al., 1983; Coan & Collingridge, 1985; Lester et al., 1988). The selectivity of CGP 37849 was indicated by its negligible interactions with binding sites for a range of other neurotransmitter receptors, including those for the quisqualate and kainate excitatory receptor sub-types, while its weak activity at strychnine-insensitive glycine binding sites provided further evidence that its primary locus of action on the NMDA rncoptor was indeed the transmitter recognition site. Additional demonstrations of its selectivity were obtained from electrophysiological studies in the hippocampus in vitro and in vivo, in which it failed to affect neuronal responses to applied quisqualate or kainate at doses which reduced or abolished responses to NMDA. With a Ki of 35 nm at [3H]-CPP binding sites, CGP 37849 thus shows a higher affinity and selectivity for the NMDA receptor recognition site than any compound described to date. NMDA receptor antagonists have been shown to exhibit anticonvulsant activity following central or systemic (i.p. or i.v.) administration to a variety of animal species (see Meldrum, 1985; Lehmann et al., 1987; 1988; Patel et al., 1988), and the oral efficacy of CGP 37849 was initially discerned from its ability to prevent maximal electroshockinduced seizures in mice; Its potency in this respect (ED50 21 mg kg-, p.o.) is within a therapeutically-useful dose-range as compared with established anticonvulsant drugs (see Meldrum & Porter, 1986), and was further augmented by esterification of the carboxyl moiety (yielding CGP 39551, ED50 4mgkg- 1, p.o.) (detailed anticonvulsant profiles for CGP 37849 and CGP 39551 will be published elsewhere). That these compounds are indeed acting centrally as NMDA receptor antagonists following oral administration was suggested by their observed enantiomeric selectivity (D-isomer active, as for all known NMDA receptor antagonists; see Watkins et al., 1990), and was more directly demonstrated by microionophoretic experiments in the rat hippocampus in vivo. At doses some 5-7 fold higher than their oral ED50s as anticonvulsants, both compounds were shown to abolish neuronal responses to NMDA, without affecting those to quisqualate, kainate or acetylcholine. The onset of action of CGP 39551 in this regard was somewhat slower than that of CGP 37489. This observation, coupled with the weaker action of CGP 39551 in vitro and current understanding of structureactivity relationships at the NMDA receptor (Fagg & Baud, 1988; Watkins et al., 1990), may indicate that the esterified compound functions as a more-readily absorbed pro-drug of CGP 37849 in vivo. However, definitive proof of the precursor-product relationship of these two substances in vivo is required to verify this hypothesis. CGP 37849 and CGP 39551 are the first competitive NMDA receptor antagonists reported to show significant central effects following oral administration in animals, and hence are candidates for the treatment of neurological disorders in man which require either acute or chronic drug administration. Whilst extensive discussion is outside the realms of this paper, it is appropriate to consider some of the issues which must be resolved before this goal can be realized. Major questions have centred on the therapeutic window of NMDA receptor antagonists (loss of muscle tone occurs at sufficient dosage; see Turski et al., 1988), their possible psychotomimetic effects (based on experience in man with noncompetitive blockers such as PCP and ketamine; see Domino & Luby, 1981; Lodge et al., 1988), and their potentially detrimental effects on learning (Morris et al., 1986; Collingridge & Bliss, 1987). In this respect, recent evidence indicates that competitive NMDA receptor antagonists may have significant advantages over non-competitive blockers (Meldrum, 1985; Ferkany et al., 1988; Leander et al., 1988; France et al., 1989; Tricklebank et al., 1989). Studies on the novel competitive NMDA receptor antagonists described here indicate, especially in the case of CGP 39551, a therapeutic window at least as great as clinically-used anticonvulsant agents (Fagg et al., 1989a; Schmutz et al., unpublished observations), no direct impairments of learning performance in the anticonvulsant dose-range (Mondadori, Buerki & Petschke, unpublished), and no generalization to the discriminative stimulus effects of ketamine or MK801 in rhesus monkeys (France & Woods, personal communication). Pending favourable toxicological profiles, they are thus leading candidates for clinical testing in man.[2] The competitive N-methyl-D-aspartate (NMDA) antagonist DL-2-amino-4-methyl-5-phosphono-3-pentenoic acid (CAS 127910-31-0, 4-methyl-APPA, CGP 37849) and its ethyl ester (CAS 127910-32-1, CGP 39551) potently block NMDA-evoked whole-cell current on mouse spinal neurones in primary dissociated cell cultures with IC50 (+/- SE) values of 189 +/- 9 nmol/l (CGP 37849) and 2100 +/- 220 nmol/l (CGP 39551), respectively. The compounds dose-dependently blocked vestibular stimulation-induced convulsions in EL mice, 2 h after oral administration, with ED50 (95% CI) values of 135 (78-236) mumol/kg (CGP 37849) and 65 (45-94) mumol/kg (CGP 39551). In male Swiss albino mice, performance in the step-through passive avoidance procedure was dose-dependently impaired with ED50 (95% CI) values of 85 (56-157) mumol/kg (CGP 37849) and 27 (18-42) mumol/kg (CGP 39551). In addition performance of these animals in the rotarod test of motor coordination was impaired, 2 h after oral administration of CGP 39551, with an ED50 (95% CI) of 142 (100-201) mumol/kg. These findings demonstrate anticonvulsant activity in these potent NMDA antagonists after oral administration with CGP 39551 possessing greater relative potency. However, the unfavourable ratio of therapeutic dose versus dose inducing memory or motor impairment supports the prevailing notion that such adverse effects of the presently available compounds preclude the use of NMDA antagonists as long-term therapies.[https://pubmed.ncbi.nlm.nih.gov/9893924/] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.2160 mL | 21.0801 mL | 42.1603 mL | |
| 5 mM | 0.8432 mL | 4.2160 mL | 8.4321 mL | |
| 10 mM | 0.4216 mL | 2.1080 mL | 4.2160 mL |