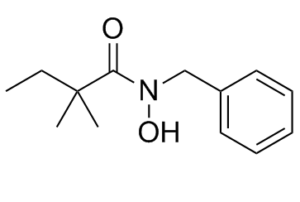
RIPA-56 is a novel, highly potent, selective, and metabolically stable inhibitor of receptor-interacting protein 1 (RIP1) with an IC50 of 13 nM. Tumor necrosis factor alpha (TNFα)-induced mortality and multiorgan damage were successfully decreased by RIPA-56. RIPA-56 is more effective in animal model studies, much more stable in vivo, and potent in both human and murine cells compared to other known RIP1 inhibitors.
Physicochemical Properties
| Molecular Formula | C13H19NO2 |
| Molecular Weight | 221.29546380043 |
| Exact Mass | 221.14 |
| Elemental Analysis | C, 70.56; H, 8.65; N, 6.33; O, 14.46 |
| CAS # | 1956370-21-0 |
| Related CAS # | 1956370-21-0 |
| PubChem CID | 121439991 |
| Appearance | White to off-white solid powder |
| LogP | 2.5 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 2 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 16 |
| Complexity | 232 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | AVYVHIKSFXVDBG-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C13H19NO2/c1-4-13(2,3)12(15)14(16)10-11-8-6-5-7-9-11/h5-9,16H,4,10H2,1-3H3 |
| Chemical Name | N-benzyl-N-hydroxy-2,2-dimethylbutanamide |
| Synonyms | RIPA-56; RIPA56; 1956370-21-0; N-benzyl-N-hydroxy-2,2-dimethylbutanamide; CHEMBL4092421; MFCD30738006; compound 92 [WO2016101885]; compound 56 [PMID: 27992216]; compound 92 (WO2016101885); RIPA 56 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | RIP1 (IC50 = 13 nM) |
| ln Vitro |
RIPA-56 shows efficient inhibition of RIP1 kinase activity, with an IC50 of 13 nM.At a 10 µM concentration, it showed no evidence of inhibiting RIP3 kinase activity. Based on the 13 nM RIP1 ADP-Glo activity, RIPA-56 does not inhibit IDO activity at a concentration of 200 µM, which is thought to have a 10,000-fold selectivity window[1].
According to the band shift results shown in Figure 3A, 56 (RIPA-56 ) can block the phosphorylation of both RIP1 and RIP3. The 56-mediated inhibition was confirmed using an antibody specific for phosphorylation on Serine227 of RIP3. 56 thus appears to inhibit the kinase activity of RIP1 and/or RIP3 or to somehow interfere with their upstream activators. The kinase activities of RIP1 and RIP3 were next monitored in vitro using an ADP-Glo assay. Compound 56 showed efficient inhibition of RIP1 kinase activity, with an IC50 of 13 nM (Figure 3B), 57-fold more potent than 1 (IC50 = 760 nM). It showed no inhibition of RIP3 kinase activity at a 10 μM concentration (Figure s2A). Several new compounds were evaluated using this RIP1 kinase activity assay. A good correlation was noted between kinase inhibition activity data and cell necrosis inhibition data (Figure s2B), indicating that RIP1 is the direct target of these new amide series inhibitors.[1] Compound 56 was also tested in IDO enzyme activity assays. Unlike 1, (8, 21, 22)56 did not inhibit IDO activity at a concentration of 200 μM, which represents an estimated 10000-fold selectivity window based on the RIP1 ADP-Glo activity of 13 nM; 56 therefore avoids the problematic off-target (anti-IDO) effect of 1. [1] |
| ln Vivo |
RIPA-56 has an impressive in vivo PK profile in mice, with a 3.1 h half-life, 22% oral bioavailability (PO), and 100% bioavailability from intraperitoneal injection (IP). RIPA-56 is very good at crossing the blood–brain barrier. RIPA-56 effectively decreased mortality and multi-organ damage brought on by tumor necrosis factor alpha (TNFα) in the SIRS mouse disease model[1]. TNFα administration can cause multiple organ dysfunction and death in mice, exhibiting many similarities with the highly lethal human SIRS disorder that can be induced by infection, trauma, etc. (4, 6, 8, 15-17) To test whether 56 (RIPA-56) has any efficacy in protecting against TNFα-induced lethality, 5.5 μg of mouse TNFα (mTNFα) (∼0.25 mg/kg body weight (BW)) was intravenously injected to C57BL/6 mice (6–8 weeks, 20–25 g) and mice were either treated with multiple-dose of 56 (0.1, 1, 3 mg/kg BW, IP, 17 min before mTNFα injection and once every 12 h) or a single dose of 56 or 1 (6 mg/kg BW, IP, 17 min before mTNFα injection). As shown in Figure 5, both multiple-dose and single-dose of 56 treatment dramatically increased the survival rate of TNFα-treated mice, showing a dose-dependent effect. Mice treated with multiple doses of 56 (3 mg/kg) or a single 6 mg/kg dose of 56 had a survival rate of 100%, much higher than the TNFα/1 group, which had a survival rate of 60%. The TNFα/vehicle-treated mice group had a 50% survival rate.[1] To test the efficacy of 56 (RIPA-56) in protecting against TNFα-induced multiorgan damage, 4 μg of mouse TNFα (mTNFα) (∼0.18 mg/kg BW) was intravenously injected into C57BL/6 mice. The levels of the mouse pro-inflammatory cytokines interleukin-6 (mIL-6) and interleukin-1β (mIL-1β) increased significantly after TNFα induction, but mice treated with 56 had lower or even normal levels of cytokines (Figure 6A). Note that mice treated with a single 6 mg/kg dose of 1 had partially decreased levels of mIL-6 and mIL-1β (Figure s4), a finding consistent with the reported protection effects of 1 on mice viability. Histological examination of hematoxylin-eosin (H&E) stained spleens of TNFα-treated mice showed that both the red pulp and the white pulp were infiltrated with many immune cells and macrophages; this pattern was also seen in the cortex and medulla of the thymus (Figure 6B).The thickness of the periarteral lymphatic sheath (PALS) also increased dramatically in TNFα-treated mice. In contrast, the spleen and thymus of mice that were additionally treated with 56 were infiltrated with only a few immune cells and macrophages and otherwise appeared similar to those of non-TNFα treated control mice. In addition to the immune organs (spleen and thymus), we also examined TNFα-induced damage to other internal organs, including the myocardium, kidneys, and pancreas. For TNFα-treated mice, the level of representive biomarkers, including serum lactate dehydrogenase (LDH), creatine kinase (CK), aspartate aminotransferase (AST), creatinine, blood urea nitrogen (BUN), and amylase, increased by 2–10-fold as compared with control mice. Organ damage could also be clearly interpreted from histological staining results, which exhibited infiltration of inflammatory cells, disordered tissues, and damaged cells. In contrast, the biomarker levels of TNFα-treated mice that were also given 56 were close to those of healthy animals and the extent of organ damage in 56-treated was limited compared to the TNFα-treated mice (Figure 6).[1] |
| Enzyme Assay |
In Vitro Kinase Activity Assay and IDO Enzyme Activity Assay [1] The RIP1 kinase assay was performed in white 384-well plate. The assay buffer contained 25 mM HEPES (pH7.2), 20 mM MgCl2, 12.5 mM MnCl2, 5 mM EGTA, 2 mM EDTA, 12.5 mM β-glycerol phosphate, and 2 mM DTT. RIP1 was first incubated with compounds or DMSO control for 15 min, then ATP/MBP substrate mixture was added to initiate the reaction. The final concentration of ATP was 50 μM and MBP 20 μM. After 90 min reaction at room temperature, the ADP-Glo reagent and detection solution were added following the technical manual. The RIP3 kinase assay conditions were almost identical to that of RIP1 assay, except the assay buffer contained 5 mM MgCl2 instead of 20 mM MgCl2 and 12.5 mM MnCl2. The luminescence was measured on PerkinElmer Enspire. The IDO enzymatic assay was conducted according to the protocols previously described with some modifications. Briefly, the reaction mixture contained 50 mM MES buffer (pH 6.5), 20 mM ascorbic acid (prepared in 0.405 M Tris, pH 8.0), 2250 U/mL catalase, 10 μM methylene blue, 200 μM L-tryptophan (l-Trp), 100 nM purified recombinant IDO, and 200 μM compounds or DMSO control per reaction. The reaction was carried out at 37 °C for 3 h. The yellow color generated from the reaction with kynurenine was measured at 321 nm using Enspire plate reader. Compounds Stability Test in Liver Microsome Assays [1] Compounds were incubated with human or mouse liver microsomes and NADPH in 0.05 M Phosphate buffer (pH = 7.4) at 37 °C for 0–60 min. The reaction was quenched, and the amount of the remaining compound was analyzed using LC-MS/MS. |
| Cell Assay |
Cell Necrosis Assay [1] Cell necrosis assay is performed in 96-well cell culture plate. Each well receives 3,000 cells, which are then cultured at 37°C over night. For 24 hours, HT-29 cells are treated with 20 ng/mL TNF, 100 nM Smac Mimetics, 20 μM z-VAD-FMK, and RIPA-56. 20 ng/mL TNFα/20 μM z-VAD-FMK, and RIPA-56 are applied to L929 cells and left on the cells for 6 hours. The Cell Titer-Glo Luminescent Cell Viability Assay kit[1] is used to calculate the cell survival ratio. Western-Blot Analysis [1] Cell pellet was collected and resuspended with lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM Na3VO4, 25 mM β-glycerol-phosphate, 0.1 mM PMSF, complete protease inhibitor cocktail, and phosphatase inhibitor cocktail (Roche). The resuspended cell pellet was incubated on ice for 30 min and centrifuged at 13000g for 10 min. The supernatants were collected for Western-blot analysis. |
| Animal Protocol |
Phamacokinetics Study in Mice [1] Following intraveneous (IV), intraperitoneal (IP), or oral administration (PO) of 56 to C57BL/6 mice (n = 3), blood was sampled through eye puncture at various time points. Compound concentrations in the plasma samples were analyzed by LC-MS/MS. Pharmacokinetic parameters were determined from individual animal data using noncompartmental analysis in phoenix 64 (winNonlin 6.3). TNFα-Induced Mice SIRS Model [1] C57BL/6 mice were used at the age of 6–8 weeks with the average body weight of 20 g and were grouped randomly (n = 6–10). Mouse TNFα was diluted in endotoxin-free PBS and injected intravenously in a volume of 0.2 mL. Different doses of 56 were IP injected before (−17 min) and after (once every 12 h) mTNFα injection unless otherwise specified, while an equal amount of solvent was injected to control mice. Mice mortality was continuously monitored every 30 min until 60 h after TNFα administration. For cytokines and biomarkers determination, blood samples were collected 12 h and 24 h after TNFα injection, respectively. |
| ADME/Pharmacokinetics |
RIPA-56 also had an impressive PK profile in mice, with a 3.1 h half-life, 22% oral bioavailability (PO), and 100% bioavailability from intraperitoneal injection (IP).[1]
Notably, RIPA-56 had a half-life of 128 min in human liver microsomal stability assays. The drug-like properties of 56 (RIPA-56) were further evaluated. 56 was tested for its ability to inhibit cytochrome P450 enzymes; it had IC50 values higher than 50 μM against CYP1A2, 2C19, 2C9, and 2D6 and higher than 3.5 μM against CYP3A4. In addition, a tissue-distribution study indicated that 56 could be delivered readily to most organs where diseases might occur (Figure s5). For example, 56 could be detected in the brains of mice following intravenous injection and exhibited strong permeability in MDCK-MDR1 cells (Papp, A–B and B–A >30 × 106 cm/s), with an efflux ratio of 0.87 (Table s3), showing great ability in transporting across the blood–brain barrier. This is attractive when considering the requirements of therapies against neurodegenerative disease and in treatment of ischemic or traumatic brain injuries in that 56 could be administrated by IV or IP injection rather than through intracerebroventricular administration. Manual patch clamp evaluation of 56 demonstrated that it did not inhibit the hERG channel, even at concentrations as high as 30 μM. Multiple-, high-strength doses of 56 (50 mg/kg, once every 3 h, for 24 h) caused no injury to mice. These results from both in vitro and in vivo studies all indicate that 56 seems to be a safe and potentially efficacious candidate drug for the treatment of necrosis-related diseases.[1] |
| References |
[1]. Discovery of a Highly Potent, Selective, and Metabolically Stable Inhibitor of Receptor-InteractingProtein 1 (RIP1) for the Treatment of Systemic Inflammatory Response Syndrome. J Med Chem. 2017 Feb 9;60(3):972-986. |
| Additional Infomation | On the basis of its essential role in driving inflammation and disease pathology, cell necrosis has gradually been verified as a promising therapeutic target for treating atherosclerosis, systemic inflammatory response syndrome (SIRS), and ischemia injury, among other diseases. Most necrosis inhibitors targeting receptor-interacting protein 1 (RIP1) still require further optimization because of weak potency or poor metabolic stability. We conducted a phenotypic screen and identified a micromolar hit with novel amide structure. Medicinal chemistry efforts yielded a highly potent, selective, and metabolically stable drug candidate, compound 56 (RIPA-56). Biochemical studies and molecular docking revealed that RIP1 is the direct target of this new series of type III kinase inhibitors. In the SIRS mice disease model, 56 efficiently reduced tumor necrosis factor alpha (TNFα)-induced mortality and multiorgan damage. Compared to known RIP1 inhibitors, 56 is potent in both human and murine cells, is much more stable in vivo, and is efficacious in animal model studies. [1] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~44 mg/mL (~198.8 mM) Ethanol: ~ 44 mg/mL (~198.8 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.75 mg/mL (12.43 mM) (saturation unknown) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.75 mg/mL (12.43 mM) (saturation unknown) in 5% DMSO + 95% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (11.30 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 4: ≥ 2.5 mg/mL (11.30 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 5: ≥ 2.5 mg/mL (11.30 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.5188 mL | 22.5938 mL | 45.1875 mL | |
| 5 mM | 0.9038 mL | 4.5188 mL | 9.0375 mL | |
| 10 mM | 0.4519 mL | 2.2594 mL | 4.5188 mL |