Physicochemical Properties
| Molecular Formula | C18H14CL2N2O3 |
| Molecular Weight | 377.2214 |
| Exact Mass | 376.038 |
| CAS # | 433238-84-7 |
| PubChem CID | 2290641 |
| Appearance | Typically exists as solid at room temperature |
| LogP | 4.4 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 3 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 25 |
| Complexity | 546 |
| Defined Atom Stereocenter Count | 0 |
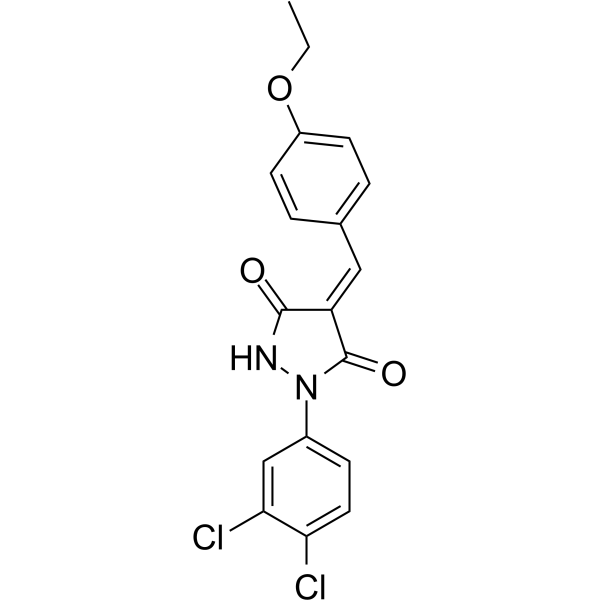
| SMILES | CCOC1=CC=C(C=C1)/C=C/2\C(=O)NN(C2=O)C3=CC(=C(C=C3)Cl)Cl |
| InChi Key | XFGORAOOZWWQKK-NTEUORMPSA-N |
| InChi Code | InChI=1S/C18H14Cl2N2O3/c1-2-25-13-6-3-11(4-7-13)9-14-17(23)21-22(18(14)24)12-5-8-15(19)16(20)10-12/h3-10H,2H2,1H3,(H,21,23)/b14-9+ |
| Chemical Name | (4E)-1-(3,4-dichlorophenyl)-4-[(4-ethoxyphenyl)methylidene]pyrazolidine-3,5-dione |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
The target of PP7 is the PB1-PB2 interface of influenza A virus polymerase. It inhibits the interaction between PB1 and PB2 subunits, with an IC50 of 2.3 μM (detected by HTRF assay for PB1-PB2 binding) and EC50 values of 0.8 μM (against H1N1 subtype), 1.1 μM (against H3N2 subtype), and 1.5 μM (against H5N1 subtype) for inhibiting influenza A virus replication [1] |
| ln Vitro |
1. Anti-influenza A virus activity: In MDCK cells infected with different influenza A virus subtypes, PP7 dose-dependently inhibited virus replication. The EC50 values were 0.8 μM (H1N1, A/PR/8/34), 1.1 μM (H3N2, A/Udorn/72), and 1.5 μM (H5N1, A/Vietnam/1203/04). At a concentration of 5 μM, PP7 reduced the viral titer (TCID50/mL) of H1N1 by 3.2 log10, H3N2 by 2.8 log10, and H5N1 by 2.5 log10 compared to the untreated infected group [1] 2. Inhibition of PB1-PB2 interaction: Using HTRF assay to detect the binding of recombinant PB1 (N-terminal fragment) and PB2 (C-terminal fragment), PP7 showed a dose-dependent inhibitory effect on their interaction with an IC50 of 2.3 μM. At 10 μM, the binding rate was reduced by over 80% compared to the control group [1] 3. Inhibition of viral RNA synthesis: In H1N1-infected A549 cells, PP7 (5 μM) reduced the levels of viral M gene RNA by 75% (detected by qPCR) and viral polymerase activity by 68% (detected by luciferase reporter assay for polymerase activity) at 24 hours post-infection [1] 4. Cytotoxicity: In MDCK and A549 cells, the CC50 (50% cytotoxic concentration) of PP7 was >20 μM (MTT assay). The selectivity index (SI = CC50/EC50) was >25 (for H1N1), >18 (for H3N2), and >13 (for H5N1), indicating low in vitro cytotoxicity [1] |
| ln Vivo |
1. Antiviral effect in H1N1-infected mice: BALB/c mice (6-8 weeks old) were intranasally infected with H1N1 virus (A/PR/8/34, 10×LD50). PP7 was administered intraperitoneally at doses of 5 mg/kg, 10 mg/kg, and 20 mg/kg, once daily for 5 days (starting 1 hour post-infection). Compared to the virus-infected control group: (1) The lung viral titer (TCID50/g) was reduced by 1.8 log10 (5 mg/kg), 2.5 log10 (10 mg/kg), and 3.0 log10 (20 mg/kg) on day 5 post-infection; (2) The survival rate of mice in the 20 mg/kg group increased from 20% (control) to 80%; (3) Lung histopathology showed reduced inflammatory cell infiltration and alveolar damage in PP7-treated groups, with the 20 mg/kg group showing a 65% reduction in lung injury score [1] 2. Effect on body weight: In the H1N1-infected mouse model, the virus-infected control group showed a 25% body weight loss on day 5 post-infection. Mice treated with 10 mg/kg and 20 mg/kg PP7 had body weight losses of 10% and 5%, respectively, indicating alleviation of virus-induced weight loss [1] |
| Enzyme Assay |
1. HTRF assay for PB1-PB2 interaction: Recombinant PB1 N-terminal fragment (labeled with His-tag) and PB2 C-terminal fragment (labeled with biotin) were prepared. The reaction system contained 50 nM PB1 fragment, 50 nM PB2 fragment, different concentrations of PP7 (0.1 μM, 0.5 μM, 1 μM, 2.3 μM, 5 μM, 10 μM), and HTRF detection reagents (streptavidin-conjugated Eu3+ cryptate and anti-His antibody-conjugated XL665). The system was incubated at room temperature for 2 hours, and the fluorescence resonance energy transfer (FRET) signal was measured at 620 nm (Eu3+ emission) and 665 nm (XL665 emission). The ratio of 665 nm/620 nm fluorescence intensity was used to calculate the inhibition rate of PP7 on PB1-PB2 binding, and the IC50 value was determined from the dose-response curve [1] 2. Influenza A virus polymerase activity assay: HEK293T cells were co-transfected with plasmids encoding influenza A virus PB1, PB2, PA, NP, and a luciferase reporter plasmid driven by the influenza virus RNA promoter. After 24 hours of transfection, cells were treated with PP7 (0.5 μM, 1 μM, 2 μM, 5 μM) or DMSO. At 48 hours post-treatment, cells were lysed, and luciferase activity was measured using a luciferase assay kit. The relative luciferase activity (compared to DMSO control) was used to evaluate the inhibitory effect of PP7 on viral polymerase activity [1] |
| Cell Assay |
1. Influenza A virus replication inhibition assay (MDCK cells): - MDCK cells were seeded into 24-well plates (1×10⁵ cells/well) and cultured overnight. Cells were pretreated with PP7 (0.1 μM, 0.5 μM, 1 μM, 2 μM, 5 μM) or DMSO for 1 hour, then infected with influenza A virus (H1N1, H3N2, or H5N1) at an MOI of 0.01. After 1 hour of adsorption, unbound virus was removed, and fresh medium containing PP7 (same concentration as pretreatment) was added. - At 48 hours post-infection, the supernatant was collected to determine viral titer by TCID50 assay (calculated using Reed-Muench method). Cells were fixed with 4% paraformaldehyde, stained with anti-influenza A nucleoprotein (NP) antibody, and the percentage of NP-positive cells was counted under a fluorescence microscope to evaluate virus replication [1] 2. Cell cytotoxicity assay (MTT): - MDCK or A549 cells were seeded into 96-well plates (5×10³ cells/well) and cultured overnight. PP7 (1 μM, 5 μM, 10 μM, 20 μM, 40 μM) or DMSO was added, and cells were incubated for 48 hours. - 20 μL of MTT solution (5 mg/mL) was added to each well, followed by 4-hour incubation at 37°C. DMSO was added to dissolve formazan crystals, and absorbance at 570 nm was measured. Cell viability was calculated as a percentage of the DMSO control group, and CC50 was determined from the dose-response curve [1] 3. Viral RNA detection (qPCR): - A549 cells were infected with H1N1 virus (MOI=0.1) and treated with PP7 (5 μM) or DMSO. At 24 hours post-infection, total RNA was extracted from cells using an RNA extraction kit. - cDNA was synthesized via reverse transcription, and qPCR was performed using specific primers for influenza A virus M gene and GAPDH (internal reference). The relative expression level of viral M gene RNA was calculated using the 2^(-ΔΔCt) method [1] |
| Animal Protocol |
- H1N1-infected BALB/c mouse model: 1. Animal selection and grouping: Female BALB/c mice (6-8 weeks old, n=10 per group) were divided into 5 groups: normal control group (no infection, no drug), virus control group (infected, intraperitoneal injection of DMSO-saline mixture), PP7 low-dose group (infected, 5 mg/kg PP7), PP7 medium-dose group (infected, 10 mg/kg PP7), and PP7 high-dose group (infected, 20 mg/kg PP7). 2. Virus infection: Mice were anesthetized with isoflurane, then intranasally inoculated with H1N1 virus (A/PR/8/34) at a dose of 10×LD50 (50 μL per mouse). 3. Drug preparation and administration: PP7 was dissolved in DMSO (final concentration 5%) and diluted with normal saline. Administration was started 1 hour post-infection, via intraperitoneal injection once daily for 5 consecutive days. 4. Sample collection and detection: (1) Body weight was measured daily for 14 days to calculate survival rate; (2) On day 5 post-infection, 5 mice per group were euthanized, and lung tissues were collected to determine viral titer (TCID50 assay) and perform histopathological analysis (H&E staining); (3) Serum was collected to detect liver function (ALT, AST) and kidney function (BUN, Cr) indicators [1] |
| Toxicity/Toxicokinetics |
1. In vitro toxicity: In MDCK and A549 cells, the CC50 of PP7 was >20 μM, and the selectivity index (SI = CC50/EC50) was >13 for all tested influenza A virus subtypes, indicating low cytotoxicity [1] 2. In vivo toxicity: In BALB/c mice treated with PP7 (5-20 mg/kg, intraperitoneal injection, 5 days), no significant changes in body weight (weight change ≤5% compared to normal control) were observed. Serum levels of ALT, AST, BUN, and Cr were within the normal range, with no obvious liver or kidney toxicity. [1] |
| References |
[1]. Identification of a novel small-molecule compound targeting the influenza A virus polymerase PB1-PB2 interface. Antiviral Res. 2017 Jan;137:58-66. |
| Additional Infomation |
1. PP7 is a novel small-molecule compound designed to target the PB1-PB2 interface of influenza A virus polymerase. Its antiviral mechanism is to block the interaction between PB1 and PB2 subunits, which is essential for the assembly and catalytic activity of the viral polymerase complex, thereby inhibiting viral RNA synthesis and replication [1] 2. PP7 shows broad-spectrum inhibitory activity against different subtypes of influenza A virus (H1N1, H3N2, H5N1) and has low in vitro and in vivo toxicity, suggesting potential as a candidate for anti-influenza A virus drug development. [1] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6510 mL | 13.2549 mL | 26.5097 mL | |
| 5 mM | 0.5302 mL | 2.6510 mL | 5.3019 mL | |
| 10 mM | 0.2651 mL | 1.3255 mL | 2.6510 mL |