PF-06869206 (PF06869206) is a novel, potent, selective and orally bioavailable inhibitor of the sodium-phosphate cotransporter NaPi2a (SLC34A1) with the potential to treat CKD. It inhibits SLC34A1 with an IC50 of 380 nM. Sodium-phosphate cotransporter 2a, or NaPi2a (SLC34A1), is a solute-carrier (SLC) transporter located in the kidney proximal tubule that reabsorbs glomerular-filtered phosphate. Inhibition of NaPi2a may enhance urinary phosphate excretion and correct maladaptive mineral and hormonal derangements associated with increased cardiovascular risk in chronic kidney disease-mineral and bone disorder (CKD-MBD). To date, only nonselective NaPi inhibitors have been described. The oral PK profile of inhibitor PF-06869206 in rodents allows for the exploration of the pharmacology of selective NaPi2a inhibition.
Physicochemical Properties
| Molecular Formula | C15H14CLF3N4O2 |
| Molecular Weight | 374.745472431183 |
| Exact Mass | 374.075 |
| CAS # | 2227425-05-8 |
| PubChem CID | 134159047 |
| Appearance | White to off-white solid powder |
| LogP | 1.9 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 8 |
| Rotatable Bond Count | 2 |
| Heavy Atom Count | 25 |
| Complexity | 543 |
| Defined Atom Stereocenter Count | 1 |
| SMILES | C12C(Cl)=C(C)NC1=C(N1CCO[C@H](CO)C1)C(C#N)=C(C(F)(F)F)N=2 |
| InChi Key | ATFQBBCQZKVZJN-QMMMGPOBSA-N |
| InChi Code | InChI=1S/C15H14ClF3N4O2/c1-7-10(16)11-12(21-7)13(23-2-3-25-8(5-23)6-24)9(4-20)14(22-11)15(17,18)19/h8,21,24H,2-3,5-6H2,1H3/t8-/m0/s1 |
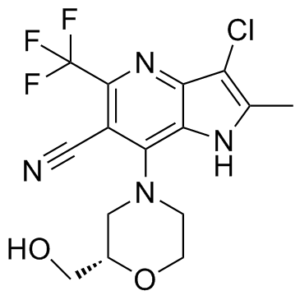
| Chemical Name | (S)-3-chloro-7-(2-(hydroxymethyl)morpholino)-2-methyl-5-(trifluoromethyl)-1H-pyrrolo[3,2-b]pyridine-6-carbonitrile |
| Synonyms | PF06869206; PF 06869206; PF-06869206 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | PF-06869206 has a good combination of isoform selectivity, 380 nM NaPi2a inhibitory activity, and acceptable water solubility (46 μM). PF-06869206 was tested for efficacy using rodent NaPi2a and NaPi2c cell lines. Comparable submicromolar activity of PF-06869206 has been seen against human, rat, and mouse NaPi2a isoforms; the IC50 values for rat and mouse NaPi2a are 0.4±0.047 μM and 0.54±0.099 μM, respectively [1]. |
| ln Vitro |
PF-06869206 has a good combination of isoform selectivity, 380 nM NaPi2a inhibitory activity, and acceptable water solubility (46 μM). PF-06869206 was tested for efficacy using rodent NaPi2a and NaPi2c cell lines. Comparable submicromolar activity of PF-06869206 has been seen against human, rat, and mouse NaPi2a isoforms; the IC50 values for rat and mouse NaPi2a are 0.4±0.047 μM and 0.54±0.099 μM, respectively [1]. PF-06869206 (also referred to as compound 6f) inhibits phosphate uptake in HEK293 cells stably expressing human NaPi2a with an IC50 of 0.38 μM. In a more physiologically relevant system, the compound (at 1 μM) inhibited phosphate uptake in primary human proximal tubule cells by an average of 32% (data from three donors), demonstrating its functional activity in cells expressing endogenous transporters (NaPi2a, NaPi2c, PiT-1, PiT-2). The inhibition was concentration-dependent. [1] |
| ln Vivo |
To ascertain whether PF-06869206 is suitable for in vivo pharmacological research, mouse PK tests were conducted. Rat and mouse clearance was found to be modest. In rats, oral bioavailability of 5 mg/kg was good; in mice, it was moderate. Both species showed a disproportionate increase in exposure at the higher oral dose of 50 mg/kg, suggesting saturation of removal. When administered intravenously (10 mg/kg), PF-06869206 has an intermediate terminal elimination half-life (t1/2=1.35 hours, 0.75 hours) in both C57BL6 mice and Wistar-Han rats (10 mg/kg). Furthermore, rat liver microsomes (RLM) have high permeability (14×10-6 cm/s) and a low clearance rate (<14 μL/min/mg; HLM=39 μL/min/mg) [1]. The provided literature does not describe specific in vivo efficacy experiments or results for PF-06869206. The manuscript states that the compound’s oral PK profile allows for the exploration of its pharmacology in vivo, but results of those efforts are stated to be communicated in a future disclosure. [1] |
| Cell Assay |
NaPi2a functional assay (Pi uptake): Phosphate uptake was measured using HEK293 cells stably expressing human NaPi2a. Cells were incubated with the test compound and ³³P-radiolabeled inorganic phosphate (Pi). After a defined incubation period, cellular uptake of radiolabeled Pi was quantified to determine the inhibitory effect of the compound. Similar assays were conducted in parallel using cell lines expressing other phosphate transporters (NaPi2b, NaPi2c, PiT-1, PiT-2) and the parental HEK293 cell line to assess selectivity. [1] Human proximal tubule cell Pi uptake assay: Primary human proximal tubule cells (from three different donors) were used. Cells were treated with the test compound and ³³P-radiolabeled Pi. The non-selective inhibitor phosphonoformic acid (PFA, 5 mM) was used as a positive control to define maximum inhibition. After incubation, cellular Pi uptake was measured to assess the compound’s ability to inhibit phosphate transport in a more native cellular context. [1] NaPi2a competition binding assay: A radioligand binding assay was developed using tritiated ([³H]) compound 6b. Membranes prepared from HEK293 cells stably expressing NaPi2a were incubated with the radioligand and increasing concentrations of the test compound (e.g., PF-06869206). The ability of the test compound to displace the radioligand was measured to determine its binding affinity (Ki). No saturable binding was observed in membranes from parental cells, confirming target-specific binding. [1] |
| Animal Protocol |
Rodent pharmacokinetic studies: Pharmacokinetic parameters were determined in Wistar-Han rats and C57BL/6 mice. For intravenous (IV) administration, PF-06869206 was formulated in a vehicle containing 10% DMSO, 30% PEG-400, and 60% water. A dose of 1 mg/kg was administered via IV injection at a volume of 2 mL/kg. For oral (PO) administration, the compound was suspended in 0.5% methylcellulose. Doses of 5 mg/kg and 50 mg/kg were administered by oral gavage at a volume of 10 mL/kg. Blood samples were collected at specified time points post-dose for the measurement of plasma drug concentrations. [1] |
| ADME/Pharmacokinetics |
In Wistar-Han rats following a 1 mg/kg IV dose, the systemic clearance (CL) was 15 mL/min/kg, the volume of distribution at steady state (Vdss) was 3.1 L/kg, and the terminal half-life (t1/2) was 4.8 hours. Following oral administration in rats, the bioavailability (F) was 87% at 5 mg/kg and 140% at 50 mg/kg, indicating supraproportional exposure increases at the higher dose suggestive of clearance saturation. The maximum plasma concentration (Cmax) was 720 ng/mL at 5 mg/kg and 6000 ng/mL at 50 mg/kg. The time to reach Cmax (tmax) was 0.75 and 1.0 hours, respectively. In C57BL/6 mice, IV clearance was 22 mL/min/kg, Vdss was 0.86 L/kg, and t1/2 was 0.75 hours. Oral bioavailability in mice was 27% at 5 mg/kg and 240% at 50 mg/kg. Cmax was 540 ng/mL and 11,000 ng/mL at the respective doses, with tmax of 0.38 and 1.5 hours. The plasma protein binding (fraction unbound, FU) was 0.0243 in rat plasma and 0.0287 in mouse plasma. The compound showed low to moderate in vitro metabolic clearance in liver microsomes (rat liver microsome clearance <14 μL/min/mg; human liver microsome clearance = 39 μL/min/mg). It exhibited good passive permeability (Papp = 15 x 10-6 cm/s in RRCK cells) and acceptable aqueous solubility (46 μM at pH 6.5). [1] |
| References |
[1]. Discovery of Orally Bioavailable Selective Inhibitors of the Sodium-Phosphate Cotransporter NaPi2a (SLC34A1). ACS Med Chem Lett. 2018 Apr 12;9(5):440-445. |
| Additional Infomation |
PF-06869206 (compound 6f) is described as the first orally bioavailable, selective inhibitor of the renal sodium-phosphate cotransporter NaPi2a (SLC34A1). It was discovered through optimization of a high-throughput screening hit (3a) to improve potency, selectivity, and drug-like properties. The compound is intended as a pharmacological tool to probe the functional effects of selective NaPi2a inhibition in vivo, with the potential therapeutic aim of promoting renal phosphate excretion, reducing serum phosphate and fibroblast growth factor 23 (FGF23) levels, and thereby potentially decreasing cardiovascular risk in patients with chronic kidney disease-mineral and bone disorder (CKD-MBD). Selective inhibition of NaPi2a over the intestinal isoform NaPi2b is highlighted as a key advantage to avoid potential risks associated with intestinal phosphate transport inhibition. [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~100 mg/mL (~266.84 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (6.67 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (6.67 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6684 mL | 13.3422 mL | 26.6845 mL | |
| 5 mM | 0.5337 mL | 2.6684 mL | 5.3369 mL | |
| 10 mM | 0.2668 mL | 1.3342 mL | 2.6684 mL |