Physicochemical Properties
| Molecular Formula | C59H100N24O17 |
| Molecular Weight | 1417.58 |
| Exact Mass | 1416.76982 |
| CAS # | 2896181-32-9 |
| PubChem CID | 172638599 |
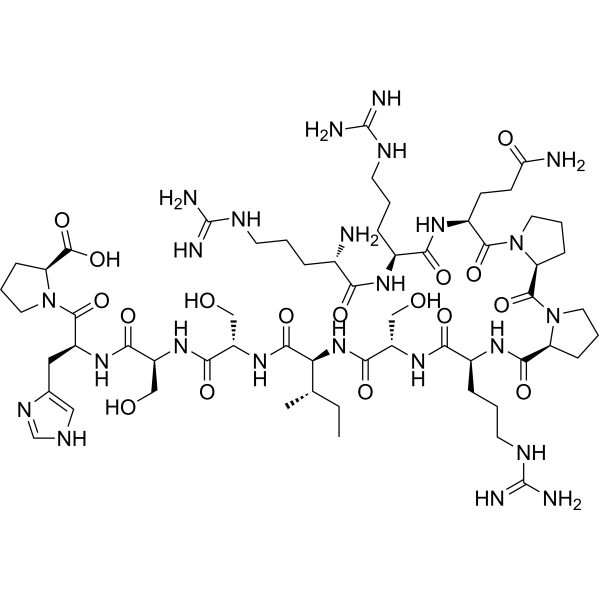
| Sequence | L-arginyl-L-arginyl-L-glutaminyl-L-prolyl-L-prolyl-L-arginyl-L-seryl-L-isoleucyl-L-seryl-L-seryl-L-histidyl-L-proline; H-Arg-Arg-Gln-Pro-Pro-Arg-Ser-Ile-Ser-Ser-His-Pro-OH |
| SequenceShortening | RRQPPRSISSHP |
| Appearance | Typically exists as solids at room temperature |
| LogP | -11.7 |
| Hydrogen Bond Donor Count | 21 |
| Hydrogen Bond Acceptor Count | 22 |
| Rotatable Bond Count | 42 |
| Heavy Atom Count | 100 |
| Complexity | 2930 |
| Defined Atom Stereocenter Count | 13 |
| SMILES | CC[C@H](C)[C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC1=CN=CN1)C(=O)N2CCC[C@H]2C(=O)O)NC(=O)[C@H](CO)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@@H]3CCCN3C(=O)[C@@H]4CCCN4C(=O)[C@H](CCC(=O)N)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CCCN=C(N)N)N |
| InChi Key | DHODDTORTQRISO-OFONGQOJSA-N |
| InChi Code | InChI=1S/C59H100N24O17/c1-3-30(2)44(52(95)79-38(27-85)49(92)78-37(26-84)48(91)76-36(24-31-25-68-29-72-31)54(97)83-23-9-15-42(83)56(99)100)80-50(93)39(28-86)77-47(90)34(12-6-20-71-59(66)67)74-51(94)40-13-7-21-81(40)55(98)41-14-8-22-82(41)53(96)35(16-17-43(61)87)75-46(89)33(11-5-19-70-58(64)65)73-45(88)32(60)10-4-18-69-57(62)63/h25,29-30,32-42,44,84-86H,3-24,26-28,60H2,1-2H3,(H2,61,87)(H,68,72)(H,73,88)(H,74,94)(H,75,89)(H,76,91)(H,77,90)(H,78,92)(H,79,95)(H,80,93)(H,99,100)(H4,62,63,69)(H4,64,65,70)(H4,66,67,71)/t30-,32-,33-,34-,35-,36-,37-,38-,39-,40-,41-,42-,44-/m0/s1 |
| Chemical Name | (2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-1-[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-amino-5-(diaminomethylideneamino)pentanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]amino]-5-oxopentanoyl]pyrrolidine-2-carbonyl]pyrrolidine-2-carbonyl]amino]-5-(diaminomethylideneamino)pentanoyl]amino]-3-hydroxypropanoyl]amino]-3-methylpentanoyl]amino]-3-hydroxypropanoyl]amino]-3-hydroxypropanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]pyrrolidine-2-carboxylic acid |
| Synonyms | Muscle homing peptide M12; Muscle homing peptide M-12; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | Proteins on the surface of muscle cells |
| ln Vitro |
M12 mediates enhanced cellular uptake of NPs in myoblasts in vitro [2] To evaluate the active targeting of PLGA-PEG-M12 NPs to muscle cells, we examined in vitro cellular uptake of PLGA-PEG NPs and PLGA-PEG-M12 NPs in C2C12 myoblasts using fluorescence microscopy. C2C12 cells were incubated with PLGA-PEG NPs or PLGA-PEG-M12 NPs encapsulating a fluorescent dye Nile red to facilitate visualization. As shown in Figure 2A, a well-dispersed red fluorescent signal originating from Nile red encapsulated PLGA-PEG NPs was detected within the cytoplasm and around the nucleus after 1 h of incubation, indicating that PLGA-PEG NPs have been rapidly taken up by C2C12 cells. The negative surface charge of NPs leads to repulsion between particles, thus preventing agglomeration and stabilizing NP dispersion inside the cells. When C2C12 cells were incubated with PLGA-PEG-M12 NPs, the intracellular red fluorescence intensity was remarkably enhanced compared to the PLGA-PEG NP treatment, suggesting that PLGA-PEG-M12 NPs were internalized by C2C12 cells to a greater extent than nontargeted NPs. Quantitative analysis showed a 2.44-fold increase in the cellular uptake efficiency of NPs after surface engineering with M12 (Figure 2B, p ≤ 0.001). To further verify the specific interaction between PLGA-PEG-M12 NPs and surface receptors of C2C12 myoblasts, we performed competitive binding experiments by preincubating C2C12 cells with free M12 solution at a concentration of 50 μM to saturate its receptors present on the cell surface prior to treatment with PLGA-PEG-M12 NPs. Our results showed that the intracellular red fluorescence intensity after free M12 pretreatment significantly decreased compared to incubation with PLGA-PEG-M12 NPs alone (p ≤ 0.001), indicating that the interaction between PLGA-PEG-M12 NPs and receptors in C2C12 cells as well as the subsequent M12 receptor-mediated endocytosis were suppressed due to ligand competition with free M12. In contrast, preincubation with M12 did not affect cellular internalization of PLGA-PEG NPs (p > 0.05). We also investigated the cellular uptake of NPs in 3T3-L1 preadipocytes to confirm the specificity of M12 receptors in muscle cells. As shown in Figures 2A and 2C, neither enhanced cellular uptake nor binding inhibition was observed in 3T3-L1 cells. These data support that M12 mediates more efficient cellular uptake of NPs in myoblasts in vitro compared to non-specific internalization. In addition to the targeting capacity of M12 to murine C2C12 myoblasts and myofibers, our preliminary validation data have also shown that M12 exhibited high binding affinity to human skeletal myoblasts (Supplementary Figure S1), suggesting the target epitope might also be present in human muscles. To investigate whether PLGA-PEG-M12 NPs could achieve active targeting to human cells, we subsequently examined in vitro cellular uptake of PLGA-PEG NPs and PLGA-PEG-M12 NPs in human skeletal myoblasts. Similar to the results observed in the C2C12 experiments, a well-dispersed red fluorescent signal arising from Nile red encapsulated NPs was detected in the cytoplasm in both treatment groups, indicating quick cellular uptake of NPs in human skeletal myoblasts (Figure 2D). Compared to PLGA-PEG NPs, the incubation of PLGA-PEG-M12 NPs resulted in a significant increase in cellular internalization, suggesting M12 mediates enhanced NP delivery to human muscle cells (Figure 2E, p ≤ 0.001). Encapsulation of VO-OHpic into PLGA-PEG-M12 NPs achieves sustained inhibition of PTEN signaling without causing cytotoxicity in vitro [2] The effects of VO-OHpic on the PTEN signaling, including protein levels of PTEN, AKT, and ribosomal S6 kinase, were determined by western blot analysis. AKT and ribosomal S6 kinase are downstream targets in the PTEN signaling pathway with inhibition of PTEN signaling leading to activation of phosphorylated AKT and ribosomal S6 kinase [27]. Our results showed that the treatment with VO-OHpic free drug or VO-OHpic encapsulated PLGA-PEG-M12 NPs at a concentration of 10 μM did not cause any significant changes in the protein level of PTEN (Supplementary Figure S2), while significantly upregulated the protein expression of pAKT (p ≤ 0.005) and pS6 (p ≤ 0.001), suggesting efficient inhibition of enzymatic activity of PTEN (Figures 4A–4C). However, we were unable to evaluate for a longer time point due to the limitation in cell viability. The expression levels of pAKT and pS6 were consistently increased when cells were incubated with VO-OHpic loaded PLGA-PEG-M12 NPs for 12 h. It was noticed that the upregulation in pAKT expression induced by VO-OHpic loaded NPs (p ≤ 0.001) was even more significant than that in the free drug group, which might be attributed to enhanced intracellular delivery of NPs to myoblasts. When the incubation time of NPs was extended to 24 h, the protein levels of pAKT (p ≤ 0.05) and pS6 (p ≤ 0.001) maintained constantly upregulated, indicating efficient and sustained inhibition of PTEN signaling achieved by VO-OHpic loaded NPs. [2] The cytotoxicity of VO-OHpic free drug and VO-OHpic loaded PLGA-PEG-M12 NPs in C2C12 myoblasts was also determined using an MTS assay. C2C12 cells were incubated with VO-OHpic free drug or VO-OHpic loaded NPs for 12, 24, and 48 h and results were normalized to the vehicle control groups. It was found that free VO-OHpic at the concentration used resulted in a decline in cell viability in a time-dependent manner (Figure 4D). Although the treatment did not cause detectable cytotoxicity within the first 12 h, the cell viability significantly decreased to 64.78% (p ≤ 0.05) and 45.73% (p ≤ 0.01) after 24 h and 48 h of incubation, respectively. In contrast, the cell viability was well maintained after the treatment with VO-OHpic loaded NPs at comparable drug concentrations throughout the entire culture period, indicating that encapsulation of VO-OHpic into PLGA-PEG-M12 NPs exhibited a great improvement in the safety of PTEN inhibitor and preserving the viability of muscle cells. Also, no obvious cytotoxicity was observed in the group treated with blank NPs, suggesting the superior biocompatibility of M12 conjugated NP delivery system. These results together provide encouraging evidence that encapsulation of VO-OHpic into PLGA-PEG-M12 NPs achieves efficient inhibition of PTEN signaling without causing cytotoxicity. |
| ln Vivo |
M12 facilitates active targeting of NPs to skeletal muscle following systemic administration in vivo [2] To assess the targeting efficiency of PLGA-PEG-M12 NPs to skeletal muscle in vivo, we examined the biodistribution of Alexa Fluor® 488 conjugated NPs (AF488-PLGA-PEG NPs and AF488-PLGA-PEG-M12 NPs) in mdx mice following intravenous injection using a fluorescence optical imaging system. The fluorescent dye AF488-NHS ester was conjugated to PLGA-PEG-NH2 through amide bond formation followed by NP preparation using the standard protocol. The experimental design representing the timeline of NP injection is shown in Figure 3A. AF488-PLGA-PEG NPs or AF488-PLGA-PEG-M12 NPs in saline at a concentration of 80 mg/kg were injected into the tail vein of mdx mice. After 6 h of administration, AF488-PLGA-PEG-M12 NPs presented an intense fluorescence signal in the whole hindlimbs, whereas only minimal accumulation of nontargeted NPs (AF488-PLGA-PEG NPs) was detected in the hindlimb muscle tissue (Figure 3B). After whole-body imaging, mice were immediately euthanized and various tissues, including the liver, kidney, spleen, heart, and skeletal muscle, were dissected for ex vivo imaging. As shown in Figure 3C, results revealed that the fluorescence signal arising from AF488-PLGA-PEG-M12 NPs was exclusively detected in the heart and skeletal muscle, including quadriceps, gastrocnemius, and diaphragm, although there was nonspecific accumulation in the liver and kidney due to active metabolism. The injection of nontargeted AF488-PLGA-PEG NPs led to a relatively homogeneous distribution of NPs in all the organs or tissues throughout the body with significantly reduced fluorescence signals in skeletal muscle compared to those in the targeted AF488-PLGA-PEG-M12 NP group. These results were consistent with the findings from the whole-body imaging, indicating the muscle-homing property of M12. Based on the biodistribution results, the fluorescence intensity of skeletal muscle was quantified for each individual muscle group and normalized to that of the liver, which is the main metabolic detoxification organ of the body. Compared to nontargeted NPs, 1.23, 1.69, 2.31, and 1.91-fold increases in the fluorescence ratio were observed in the heart, quadriceps, gastrocnemius, and diaphragm, respectively in the mice injected with AF488-PLGA-PEG-M12 NPs (Figure 3D). The difference in gastrocnemius muscle between two groups was statistically significant (p ≤ 0.05). The data reveal that the conjugation of M12 achieves a selective enhancement in total NP localization in muscle tissues with minimizing the undesired accumulation in off-target organs. We subsequently sectioned the gastrocnemius muscle to further determine the distribution of NPs within the tissue. As shown in Figure 3E, green fluorescence signals arising from NPs were detected only in limited regions in muscle sections of AF488-PLGA-PEG NP treated mice. The intensity and area of fluorescence signals substantially increased in the section collected from the mice injected with AF488-PLGA-PEG-M12 NPs compared to those in the AF488-PLGA-PEG NP group, demonstrating M12-mediated NP accumulation in the gastrocnemius muscle. Quantitative analysis further confirmed a 1.7-fold increase in the intensity of total AF488 fluorescence in muscle sections in the M12 conjugated NP group compared to that acquired in the nontargeted NP group (Figure 3F, p ≤ 0.005). Interestingly, it was observed that a portion of AF488-PLGA-PEG-M12 NPs were localized inside myofibers within the time intervals analyzed, whereas fluorescence signals were barely seen within myofibers in the AF488-PLGA-PEG NP group (Figure 3E). Quantification results showed that the intensity of AF488 fluorescence within myofibers in the AF488-PLGA-PEG-M12 NP group was approximately 2.1-fold higher than that in the AF488-PLGA-PEG NP group (Figure 3G, p ≤ 0.01), suggesting that M12 might facilitate the binding of NPs to myofibers and subsequent permeation across the myofiber membrane. Nevertheless, the basal lamina and myofiber sarcolemma posed a significant delivery barrier for NPs as the majority of fluorescent NPs were retained in the extracellular matrix in both formation groups at the time point examined (i.e. 6 h post injection). |
| Cell Assay |
Cell culture and cellular uptake of NPs [2] C2C12 myoblasts, 3T3-L1 preadipocytes, and human skeletal myoblasts were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and 1% antibiotics (penicillin and streptomycin) at 37 °C, 5% CO2, and 95% relative humidity. C2C12 myoblasts, 3T3-L1 preadipocytes, and human skeletal myoblasts were seeded onto 12-well plates at a density of 5 × 104 cells per well and cultured for 24 h. After washing with PBS, cells were incubated with DMEM containing Nile red encapsulated NPs at a final concentration of 0.1 mg/mL for 1 h. Subsequently, cell nuclei were stained with Hoechst 33342 (1 μg/mL in PBS) at for 10 min at 37 °C following by repeated washing with PBS to remove remaining NPs and dead cells. Fluorescent images were captured using a CoolSnap HQ charge coupled-device camera equipped on a Leica DM 6000B microscope with a × 20 objective. Cells without any NP treatment were used as negative control. For competitive inhibition studies, C2C12 myoblasts and 3T3-L1 preadipocytes were preincubated with free M12 solution in DMEM at a concentration of 50 μM for 1 h at 37 °C, followed by the same procedure as described above. The corrected total cell fluorescence was quantified by measuring integrated density with background subtraction using ImageJ software. 200–300 cells in total from three individual repeats were analyzed for each group. Western blot analysis of PTEN downstream targets [2] C2C12 myoblasts were seeded onto 6-well plates at a density of 3 × 104 cells per well. After 24 h of culture, free VO-OHpic solution or VO-OHpic loaded PLGA-PEG-M12 NP suspension was added into the culture medium at a final VO-OHpic concentration of 10 μM and cells were further cultured for 12 or 24 h. The cells treated with DMSO vehicle only and blank PLGA-PEG-M12 NPs without encapsulating drugs were used as negative controls. Total protein was extracted from cells using RIPA buffer containing 25 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate and 0.1% SDS. Protein concentrations were determined by Pierce BCA Protein Assay Reagent. Proteins were separated by SDS-PAGE and transferred to a polyvinylidene fluoride membrane. The membrane was then blocked with 5% fat-free milk for 1 h at room temperature and incubated with primary antibodies in 5% milk overnight at 4°C as well as secondary antibodies for 1 h at room temperature. Primary antibodies, including pAKT (1:2000 dilution), AKT (1:2000 dilution), pS6 (1:2000 dilution), S6 (1:2000 dilution), and β-Actin (1:5000 dilution), were used. HRP-conjugated secondary antibodies, including anti-rabbit and anti-mouse IgG, were also used at a dilution of 1:10,000. A luminol reagent for enhanced chemiluminescence detection of western blots was employed and signals were detected with a FluorChem R imaging system. Assessment of cell viability [2] In vitro cell viability after incubation with free VO-OHpic or VO-OHpic loaded PLGA-PEG-M12 NPs was determined using an MTS assay, which is based on the mitochondrial conversion of a tetrazolium salt. C2C12 cells were seeded onto 96-well plates at a density of 1 × 103 cells per well and cultured for 24 h. Blank NPs, VO-OHpic loaded NPs, or free VO-OHpic solutions were then added to the cells in culture medium at a final drug concentration of 10 μM and cells were further cultured for 12, 24, or 48 h. Subsequently, 20 μL of CellTiter 96® AQueous one solution reagent was added to each well in 100 μL of culture medium and incubated at 37 °C in a humidified, 5% CO2 atmosphere for 2 h. The absorbance was measured using a Tecan Spark™ 10M microplate reader at a wavelength of 490 nm with background subtraction at 680 nm. Cells incubated with 0.1% Triton X-100 and DMSO vehicle only in culture medium served as positive and negative controls, respectively. |
| Animal Protocol | Fluorescent AF 488 conjugated NPs (AF488-PLGA-PEG NPs or AF488-PLGA-PEG-M12 NPs) with a single dose of 80 mg/kg body weight were injected via the tail vein (intravenous injection) into mdx mice. Mice were anesthetized with Ketamine administered by intraperitoneal injection at 6 h post injection. Hairs on both hindlimbs were removed and the exposure parameters were adjusted using a control mouse without any treatment to avoid background autofluorescence. Images were taken with the Ami optical imaging system. After whole-body scanning (6 h post injection), mice were immediately euthanized and multiple tissues, including the liver, kidney, spleen, heart, quadriceps, gastrocnemius, and diaphragm, were collected, followed by the ex vivo imaging. The fluorescent images were analyzed by the Living Image Software and quantification of fluorescence ratio of muscle to liver was performed by dividing the fluorescence intensity of liver with that of each muscle tissue. [2] |
| References |
[1]. Nanomaterial for Skeletal Muscle Regeneration. Tissue Eng Regen Med. 2022 Apr;19(2):253-261. [2]. Polymeric nanoparticles functionalized with muscle-homing peptides for targeted delivery of phosphatase and tensin homolog inhibitor to skeletal muscle. Acta Biomater. 2020 Dec:118:196-206. |
| Additional Infomation |
Skeletal muscle has an innate regenerative capacity to restore their structure and function following acute damages and injuries. However, in congenital muscular dystrophies, large volumetric muscle loss, cachexia, or aging, the declined regenerative capacity of skeletal muscle results in muscle wasting and functional impairment. Recent studies indicate that muscle mass and function are closely correlated with morbidity and mortality due to the large volume and location of skeletal muscle. However, the options for treating neuromuscular disorders are limited. Biomedical engineering strategies such as nanotechnologies have been implemented to address this issue.In this review, we focus on recent studies leveraging nano-sized materials for regeneration of skeletal muscle. We look at skeletal muscle pathologies and describe various proof-of-concept and pre-clinical studies that have used nanomaterials, with a focus on how nano-sized materials can be used for skeletal muscle regeneration depending on material dimensionality.Depending on the dimensionality of nano-sized materials, their application have been changed because of their different physical and biochemical properties.Nanomaterials have been spotlighted as a great candidate for addressing the unmet needs of regenerative medicine. Nanomaterials could be applied to several types of tissues and diseases along with the unique characteristics of nanomaterials. However, when confined to muscle tissue, the targets of nanomaterial applications are limited and can be extended in future research. [1] hosphatase and tensin homolog (PTEN) antagonizes muscle growth and repair, and inhibition of PTEN has been shown to improve the pathophysiology and dystrophic muscle function in a mouse model of Duchenne muscular dystrophy (DMD). However, conventional pharmacological delivery of PTEN inhibitors carries a high risk of off-target side effects in other non-muscle organs due to broad targeting spectrums. Here we report a muscle-targeted nanoparticulate platform for cell-specific delivery of a PTEN inhibitor. Poly(lactide-co-glycolide)-b-poly(ethylene glycol) nanoparticles (NPs) are functionalized with a muscle-homing peptide M12 to promote the selective uptake by muscle cells/tissue in vitro and in vivo. Moreover, the NPs are formulated to slowly release the PTEN inhibitor, preventing cytotoxicity associated with direct exposure to the drug and facilitating sustained inhibition of PTEN. This advanced delivery approach taking advantages of polymeric nanomaterials and muscle-homing peptides opens a new avenue for the development of long-term therapeutic strategies in DMD treatment. [2] Pharmacological inhibition of phosphatase and tensin homolog (PTEN) has been demonstrated to improve muscle function in a mouse model of Duchenne muscular dystrophy (DMD), but translation of this approach into clinical settings remains challenging due to potential risks of off-target side effects. Herein, we developed a nanoparticulate platform, consisting of poly(lactide-co-glycolide)-b-poly(ethylene glycol) and a muscle-homing peptide M12, for cell-specific delivery of a PTEN inhibitor. M12 facilitates the cellular internalization of nanoparticles in myoblasts and their selective localization in skeletal muscle. Moreover, the slowly released drug from nanoparticles reduces its cytotoxicity and achieves sustained PTEN inhibition. This advanced delivery approach taking advantages of nanomaterials and targeting peptides opens a new avenue for the development of long-term therapeutic strategies in DMD treatment.[2] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 0.7054 mL | 3.5271 mL | 7.0543 mL | |
| 5 mM | 0.1411 mL | 0.7054 mL | 1.4109 mL | |
| 10 mM | 0.0705 mL | 0.3527 mL | 0.7054 mL |