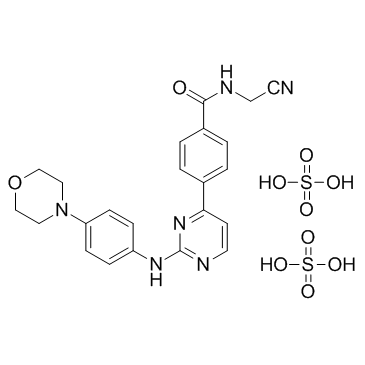
Momelotinib sulfate (CYT387) is a novel and potent inhibitor of JAK1/JAK2 with with potential antitumor and anti-inflammatory activity. It inhibits JAK1/2 with IC50 of 11 nM/18 nM, 10-fold selectivity versus JAK3 (IC50=155 nM). Momelotinib (Ojjaara) was approved in 2023 by FDA for treating Myelofibrosis in adults with anaemia.
Physicochemical Properties
| Molecular Formula | C23H22N6O2.2[H2O4S] |
| Molecular Weight | 610.61674 |
| Exact Mass | 610.115 |
| CAS # | 1056636-06-6 |
| Related CAS # | Momelotinib;1056634-68-4;Momelotinib Mesylate;1056636-07-7 Momelotinib sulfate;1056636-06-6; 1380317-28-1 (HCl) |
| PubChem CID | 66576992 |
| Appearance | Light yellow to yellow solid powder |
| LogP | 4.362 |
| Hydrogen Bond Donor Count | 6 |
| Hydrogen Bond Acceptor Count | 15 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 41 |
| Complexity | 696 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | CS(=O)(=O)O.C1COCCN1C2=CC=C(C=C2)NC3=NC=CC(=N3)C4=CC=C(C=C4)C(=O)NCC#N |
| InChi Key | XJGPMRGWDSQVTN-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C23H22N6O2.2H2O4S/c24-10-12-25-22(30)18-3-1-17(2-4-18)21-9-11-26-23(28-21)27-19-5-7-20(8-6-19)29-13-15-31-16-14-29;2*1-5(2,3)4/h1-9,11H,12-16H2,(H,25,30)(H,26,27,28);2*(H2,1,2,3,4) |
| Chemical Name | N-(cyanomethyl)-4-[2-(4-morpholin-4-ylanilino)pyrimidin-4-yl]benzamide;sulfuric acid |
| Synonyms | Ojjaara; momelotinib Mesylate; 1056636-07-7; CYT387 Mesylate; CYT387 (Mesylate); N-(cyanomethyl)-4-[2-(4-morpholin-4-ylanilino)pyrimidin-4-yl]benzamide;methanesulfonic acid; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | JAK1/2 | ||
| ln Vitro | Momelotinib sulfate, also known as CYT387 sulfate salt, suppresses the growth of Ba/F3-JAK2V617F and human erythroleukemia (HEL) cells (IC50=1.5 μM) or Ba/F3-MPLW515L cells (IC50=200 nM). However, it exhibits significantly less activity against K562 cells that harbor BCR-ABL (IC50=58 μM) and MV4-11 cells that harbor FLT3 mutation (IC50=3 μM). With an IC50 value of 1.4 μM, the proliferation of parental Ba/F3 cells (Ba/F3-wt) stimulated with IL-3 is suppressed, in keeping with the known function of IL-3-dependent signaling in the parental cell line[1]. | ||
| ln Vivo | Over the course of eight weeks, momelotinib sulfate (CYT387 sulfate salt), at twice the dose employed in the illness model (50 and 100 mg/kg), has little to no effect on peripheral blood counts. With a half-life of roughly two hours, the median plasma peak concentrations are 7.1 μM for the lower dose and 32.1 μM for the higher dose. Twelve-hour trough values for the 25 mg/kg and 50 mg/kg doses, respectively, are 10nM and 900nM. The cohort's average white blood cell counts and hematocrit values at day 34 post-transplantation were more than one standard deviation above the usual range for Balb/c mice. Six mice are now sacrificed and put through an autopsy. Treatment is started twice daily by oral gavage (12 mice per treatment group) with 25 mg/kg Momelotinib sulfate (CYT387 sulfate salt), 50 mg/kg Momelotinib sulfate (CYT387 sulfate salt), or vehicle for the remaining animals. Within 6 days of treatment beginning, both dose cohorts show a sharp reduction in white cell counts, and after 20 days, there is a noticeable decline in hematocrit[2]. Momelotinib sulfate (CYT387 sulfate salt) has an apparent half life of 2.4 hours, a quantitative absolute oral bioavailability, and high plasma concentrations (Cmax= 40.4 μM; Tmax= 4 h) following oral administration. The low blood clearance of momelotinib sulfate (CYT387 sulfate salt) (6.3 mL/min/kg) and consequently poor susceptibility to hepatic first pass metabolism are probably contributing factors to the drug's high oral bioavailability[3]. | ||
| Animal Protocol |
|
||
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Momelotinib is rapidly absorbed following oral administration with a bioavailability of 97%. The mean (%CV) steady-state Cmax is 479 ng/mL (61%), and the mean (%CV) AUC is 3,288 ng x h/mL (60%) at the maximum recommended dosage. Momelotinib exposure (i.e., Cmax and AUC) increases dose proportionally from 100 mg to 300 mg (0.5 to 1.5 times the maximum recommended dosage), but less than dose-proportional at doses from 400 mg to 800 mg (two to four times the maximum recommended dosage). There is no clinically significant accumulation. The Tmax at steady state is two hours (Q1: 1 hour; Q3: 3 hours) post-dose. No clinically significant differences in momelotinib pharmacokinetics were observed following administration of either a high-fat meal (800 kcal; 50% fat) or low-fat meal (400 kcal; 20% fat) in healthy subjects. Momelotinib is primarily eliminated in feces and, to a lesser extent, in urine. Following a single oral dose of radiolabeled momelotinib in healthy subjects, about 69% of the total radioactive dose was recovered in fecesm with M14 accounting for 21.4% of the dose, momelotinib and M21 each accounting for 13%, and other 12 metabolites accounting for the remaining 22%. About 28% of radioactivity was recovered in urine, with M21 being the major species. The mean (%CV) apparent volume of distribution at steady-state is 984 L (118%). The mean (%CV) clearance is clearance is 103 L/h (87%). Metabolism / Metabolites Momelotinib is metabolized by multiple cytochrome P450 (CYP) enzymes, including CYP3A4 (36%), CYP2C8 (19%), CYP2C9 (17%), CYP2C19 (19%), and CYP1A2 (9%). M21 is initially formed via oxidation of the morpholine ring by the same CYP enzymes, followed by metabolism via aldehyde oxidase. M21 is a major metabolite in humans that retains approximately 40% of the pharmacological activity of the parent. The mean ratio of M21 to momelotinib for AUC ranged from 1.4 to 2.1. Momelotinib can undergo amide hydrolysis, N-dealkylation, nitrile hydrolysis, nitrile oxidation, and glucuronidation. Biological Half-Life The elimination half-life of momelotinib and the M21 metabolite is four to eight hours. Oral bioavailability in rats: Male Sprague-Dawley rats (250–300 g) received Momelotinib (LM1149; CYT387; CYT11387) via oral gavage (10 mg/kg) or intravenous injection (2 mg/kg): - Oral bioavailability = 50%; - Oral administration: Cmax = 3.1 μg/mL (Tmax = 1.6 h), terminal half-life (t1/2) = 4.3 h, AUC0-24h = 17.2 μg·h/mL; - Intravenous administration: Cmax = 7.9 μg/mL, t1/2 = 3.9 h, AUC0-∞ = 34.4 μg·h/mL [1] - Plasma protein binding: In human plasma, Momelotinib (LM1149; CYT387; CYT11387) had a protein binding rate of 93% (measured by equilibrium dialysis at 37°C) [1] - Tissue distribution in MPN mice: Oral Momelotinib (LM1149; CYT387; CYT11387) (60 mg/kg) in MPN mice resulted in bone marrow concentration of 4.8 μg/g and spleen concentration of 4.5 μg/g at 2 h post-administration, ~1.5-fold of plasma concentration (3.2 μg/mL) [3] |
||
| Toxicity/Toxicokinetics |
Hepatotoxicity In the published preregistration clinical trials of momelotinib, rates of serum ALT or AST elevations ranged from 21% to 31% and were above 5 times the upper limit of normal (ULN) in 0.5% to 2.0%, and above 20 times ULN in 0.5%. Two of 448 momelotinib treated patients evaluated in the safety cohort developed clinically apparent, but self-limiting liver injury with jaundice. A third patient developed liver injury with jaundice that appeared to be due to reactivation of hepatitis B. The liver injury was typically hepatocellular without immune allergic or autoimmune features, arising after 2 to 4 months of therapy, and resolving soon after drug discontinuation. Peak ALT elevations ranged from 308 to 1178 U/L and peak bilirubin from 2.3 to 7.0 mg/dL. There were no deaths from hepatic failure. Since its approval and more widespread clinical use, there have been no further reports of serum enzyme or bilirubin elevations or instances of clinically apparent liver injury, but it has been available for a limited time only. Likelihood score: D (possible cause of clinically apparent liver injury including reactivation of hepatitis B). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the clinical use of momelotinib during breastfeeding. Because momelotinib is 91% bound to plasma proteins, the amount in milk is likely to be low. The manufacturer recommends that breastfeeding be discontinued during momelotinib therapy and for at least 1 week after the last dose. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Momelotinib is 91% bound to plasma proteins in healthy volunteers. Rodent repeat-dose toxicity: Male/female Sprague-Dawley rats (n=4/sex/group) received Momelotinib (LM1149; CYT387; CYT11387) (5/30/100 mg/kg, oral, daily) for 28 days: - No mortality; no-observed-adverse-effect level (NOAEL) = 30 mg/kg; - At 100 mg/kg: Mild thrombocytopenia (platelet count reduced by 20% vs. control), no histopathological changes in liver or kidneys; serum ALT/AST/creatinine unchanged [1] - In vivo safety in MPN mice: Momelotinib (LM1149; CYT387; CYT11387) (up to 60 mg/kg, oral, 28 days) caused ≤4% body weight loss, no overt toxicity (e.g., lethargy, diarrhea), and normal serum creatinine/BUN levels [3] - In vitro normal cell safety: Human PBMCs treated with Momelotinib (LM1149; CYT387; CYT11387) (≤10 μM) for 72 h showed >85% viability (MTT assay), with no significant apoptosis [1] |
||
| References |
[1]. CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia, 2009, 23(8), 1441-1445. [2]. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood, 2010, 115(25), 5232-5240. [3]. Phenylaminopyrimidines as inhibitors of Janus kinases (JAKs). Bioorg Med Chem Lett. 2009 Oct 15;19(20):5887-92. |
Solubility Data
| Solubility (In Vitro) |
DMSO : ~220 mg/mL (~360.29 mM) H2O : ~100 mg/mL (~163.77 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 5.5 mg/mL (9.01 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 55.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 5.5 mg/mL (9.01 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 55.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 5.5 mg/mL (9.01 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 55.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 100 mg/mL (163.77 mM) in PBS (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.6377 mL | 8.1884 mL | 16.3768 mL | |
| 5 mM | 0.3275 mL | 1.6377 mL | 3.2754 mL | |
| 10 mM | 0.1638 mL | 0.8188 mL | 1.6377 mL |