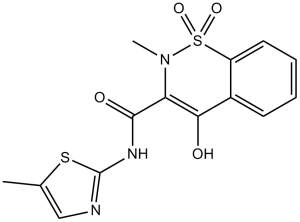
Meloxicam (Parocin; Movicox; Meloxicamum; Mobic; Mobicox; reumoxicam; Movalis; Uticox), an approved medication used to treat pain and inflammation in rheumatic arthritis and osteoarthritis, is a potent non-steroidal anti-inflammatory drug (NSAID) which acts as a selective COX inhibitor with IC50s of 0.49 µM and 36.6 µM for COX-2 and COX-1, respectively. I is used to relieve pain and fever effects. Studies suggest that Meloxicam is Cox-2 preferential, therefore it will probably not display a lower gastrointestinal toxicity than non-selective anti-inflammatory agents. This compound has been shown to also inhibit prostanoid synthesis in inflammatory cells. It shows potent anti-inflammatory, antipyretic, and analgesic effects with low gastrointestinal toxicity in animal models.
Physicochemical Properties
| Molecular Formula | C14H13N3O4S2 | |
| Molecular Weight | 351.4 | |
| Exact Mass | 351.034 | |
| CAS # | 71125-38-7 | |
| Related CAS # | Meloxicam-d3;942047-63-4;Meloxicam-d3-1;1227358-55-5;Meloxicam sodium;71125-39-8;Meloxicam-13C,d3;1309936-00-2 | |
| PubChem CID | 54677470 | |
| Appearance | Light yellow to green yellow solid powder | |
| Density | 1.6±0.1 g/cm3 | |
| Boiling Point | 581.3±60.0 °C at 760 mmHg | |
| Melting Point | 255ºC | |
| Flash Point | 305.4±32.9 °C | |
| Vapour Pressure | 0.0±1.7 mmHg at 25°C | |
| Index of Refraction | 1.735 | |
| LogP | 3.35 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 7 | |
| Rotatable Bond Count | 2 | |
| Heavy Atom Count | 23 | |
| Complexity | 628 | |
| Defined Atom Stereocenter Count | 0 | |
| InChi Key | DWMREKMVXIFPFM-ACCUITESSA-N | |
| InChi Code | InChI=1S/C14H13N3O4S2/c1-8-7-15-14(22-8)16-13(19)11-12(18)9-5-3-4-6-10(9)23(20,21)17(11)2/h3-7,19H,1-2H3,(H,15,16)/b13-11+ | |
| Chemical Name | (E)-3-(hydroxy((5-methylthiazol-2-yl)amino)methylene)-2-methyl-2H-benzo[e][1,2]thiazin-4(3H)-one 1,1-dioxide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Meloxicam is a selective cyclooxygenase-2 (COX-2) inhibitor. In in vitro assays using human recombinant COX-1 and COX-2, it exhibited high selectivity for COX-2 with an IC₅₀ of 0.08 μM, while showing weak inhibition of COX-1 with an IC₅₀ of 36 μM (selectivity ratio COX-1/COX-2 = 450) [1] - In canine mammary carcinoma cells (CF41.Mg), Meloxicam indirectly downregulates matrix metalloproteinases (MMP-2 and MMP-9) via COX-2 inhibition [2] - In inflamed mouse paw tissues, Meloxicam inhibits sorbitol dehydrogenase (SDH) activity, a key enzyme in the polyol pathway associated with inflammatory oxidative stress [3] |
| ln Vitro |
Compound 5, meloxicam, is a non-steroidal anti-inflammatory drug that suppresses COX activity. Its IC50 values for COX-2 and COX-1 are 0.49 µM and 36.6 µM, respectively[1]. At 0.25–25 µg/mL, meloxicam does not exhibit any cytotoxicity on MDCK or CF41.Mg tumor cells, but it suppresses COX+ tumor cells. Additionally, doxorubicin and meloxicam do not work synergistically on CF41.Mg cells. Meloxicam (0.25 µg/mL) inhibits the migration and invasion of CF41.Mg cells, reduces the production of MMP-2, and increases β-catenin phophorylation in CF41.Mg cells; however, it has no effect on apoptosis in CF41.Mg cells[2]. COX inhibition and prostaglandin reduction: In human whole blood assays, Meloxicam (0.01-10 μM) concentration-dependently inhibited COX-2-mediated (LPS-induced) PGE₂ production, achieving 90% inhibition at 0.2 μM; it had minimal effect on COX-1-mediated (A23187-induced) PGE₂ production even at 10 μM (inhibition < 15%) [1] - Canine mammary carcinoma cell regulation: In CF41.Mg cells, Meloxicam (5-40 μM) dose-dependently suppressed migration and invasion: - Transwell migration assay: Migrated cell number reduced by 32%, 55%, 72% at 5, 20, 40 μM vs. control; - Matrigel invasion assay: Invasive cell number reduced by 28%, 51%, 68% at 5, 20, 40 μM vs. control; - Western blot: MMP-2 and MMP-9 protein levels reduced by 35%, 58%, 70% (MMP-2) and 30%, 52%, 65% (MMP-9) at 5, 20, 40 μM vs. control; - MTT assay: No significant effect on cell viability (<10% reduction at 40 μM, 72 hours) [2] - Sorbitol dehydrogenase inhibition: In homogenates of inflamed mouse paw tissues, Meloxicam (1-20 μM) inhibited SDH activity: at 20 μM, SDH activity was reduced by 48% vs. vehicle control (measured via NADH formation assay) [3] |
| ln Vivo |
In mice, paw liking time is considerably reduced by meloxicam (10 mg/kg) alone or in combination with rutin on the first day by 55% and 49%, respectively, compared to the formalin-treated group; however, the combination does not significantly reduce time on the third day. Additionally, meloxicam alone or in combination with rutin reduces MDA contents, activates liver SOD activities, lowers relative liver weights, inhibits IL-1β content, and considerably lowers the number of positive caspase-3 immunoreactive cells in mice[3]. Mouse paw inflammation model: In male Swiss mice with carrageenan-induced paw edema (0.1 mL of 1% carrageenan, subplantar injection), oral administration of Meloxicam (1, 3, 10 mg/kg) dose-dependently reduced inflammation: - Paw volume at 4 hours post-carrageenan: reduced by 25%, 48%, 65% at 1, 3, 10 mg/kg vs. vehicle (plethysmometry); - Paw SDH activity: reduced by 22%, 40%, 55% at 1, 3, 10 mg/kg vs. vehicle; - Serum TNF-α and IL-6 levels: reduced by 30%, 52%, 68% (TNF-α) and 28%, 48%, 62% (IL-6) at 1, 3, 10 mg/kg vs. vehicle (ELISA) [3] |
| Enzyme Assay |
COX-1/COX-2 activity assay (from Reference [1]): Human recombinant COX-1 (ovarian source) and COX-2 (insect cell-expressed) were suspended in 50 mM Tris-HCl buffer (pH 8.0) containing heme (1 μM) and glutathione (1 mM). Serial concentrations of Meloxicam (0.001-100 μM) were added, followed by arachidonic acid (10 μM) as substrate. The reaction was incubated at 37°C for 15 minutes and stopped with 1 M HCl. PGE₂ production was measured by competitive radioimmunoassay (RIA) with [³H]-PGE₂. IC₅₀ values were calculated via non-linear regression of PGE₂ inhibition vs. Meloxicam concentration [1] - Sorbitol dehydrogenase activity assay (from Reference [3]): Inflamed mouse paw tissues were homogenized in 50 mM phosphate buffer (pH 7.4) and centrifuged (10,000×g, 10 minutes) to collect supernatant. Meloxicam (1-20 μM) was added to the supernatant, followed by sorbitol (10 mM) and NAD⁺ (0.5 mM) as substrates. SDH activity was quantified by measuring NADH formation (absorbance at 340 nm) over 30 minutes. Activity was expressed as nmol NADH/min/mg protein [3] |
| Cell Assay |
Canine mammary carcinoma cell migration assay (from Reference [2]): CF41.Mg cells (5×10⁴ cells/well) were seeded in upper Transwell chambers (8 μm pore size) with serum-free medium containing Meloxicam (5-40 μM); lower chambers contained medium with 10% FBS. After 24 hours, non-migrated cells on the upper membrane were removed, migrated cells on the lower membrane were fixed with 4% paraformaldehyde, stained with 0.1% crystal violet, and counted under a microscope (5 fields/well) [2] - Canine mammary carcinoma cell invasion assay (from Reference [2]): Matrigel (100 μL/well) was coated on upper Transwell chambers and polymerized at 37°C for 2 hours. CF41.Mg cells (1×10⁵ cells/well) in serum-free medium with Meloxicam (5-40 μM) were seeded into the upper chamber; lower chambers contained 10% FBS medium. After 48 hours, invasive cells were fixed, stained, and counted as described in the migration assay [2] - MMP Western blot assay (from Reference [2]): CF41.Mg cells were treated with Meloxicam (5-40 μM) for 48 hours, lysed in RIPA buffer, and proteins were separated by SDS-PAGE. Western blot was performed using anti-MMP-2 and anti-MMP-9 antibodies (GAPDH as loading control). Band intensities were quantified using ImageJ software [2] |
| Animal Protocol |
Mice, horse, and dogs Mouse paw edema protocol (from Reference [3]): Male Swiss mice (25-30 g) were randomized into 4 groups (n=8/group): - Vehicle group: 0.5% carboxymethyl cellulose (10 mL/kg, oral); - Meloxicam 1 mg/kg group: 1 mg/kg Meloxicam (dissolved in 0.5% carboxymethyl cellulose, 10 mL/kg, oral); - Meloxicam 3 mg/kg group: 3 mg/kg Meloxicam (same solvent/volume, oral); - Meloxicam 10 mg/kg group: 10 mg/kg Meloxicam (same solvent/volume, oral); Thirty minutes after drug administration, 0.1 mL of 1% carrageenan (dissolved in 0.9% saline) was injected subplantarly into the right hind paw. Paw volume was measured at 0, 1, 2, 4, 6 hours post-carrageenan using a plethysmometer. At 6 hours, mice were euthanized: paw tissues were collected for SDH activity assay; blood was collected for serum TNF-α/IL-6 measurement (ELISA) [3] |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion The absolute bioavailability oral capsules after a dose was 89% in one pharmacokinetic study. Cmax was reached 5–6 hours after administration of a single dose given after the first meal of the day. The Cmax doubled when the drug was administered in the fasting state. Despite this, meloxicam can be taken without regard to food, unlike many other NSAIDS. Meloxicam formulated for instillation with [bupivacaine] produced varied systemic measures following a single dose of varying strength. In patients undergoing bunionectomy, 1.8 mg of meloxicam produced a Cmax of 26 ± 14 ng/mL, a median Tmax of 18 h, and an AUC∞ of 2079 ± 1631 ng\\h/mL. For a 9 mg dose used in herniorrhaphy, the corresponding values were 225 ± 96 ng/mL, 54 h, and the AUC∞ was not reported. Lastly, a 12 mg dose used in total knee arthroplasty produced values of 275 ± 134 ng/mL, 36 h, and 25,673 ± 17,666 ng\\h/mL. Meloxicam is mainly eliminated through metabolism. Its metabolites are cleared through renal and fecal elimination. Less than <0.25% of a dose is eliminated in the urine as unchanged drug. About 1.6% of the parent drug is excreted in the feces. The volume of distribution of meloxicam is 10-15L. Because of its high binding to albumin, it is likely to be distributed in highly perfused tissues, such as the liver and kidney. Meloxicam concentrations in synovial fluid, measured after an oral dose, is estimated at 40% to 50% of the concentrations measured in the plasma. This drug is known to cross the placenta in humans. After an oral dose, the total clearance of meloxicam is 0.42–0.48 L/h. The FDA label indicates a plasma clearance from 7 to 9 mL/min. No dose changes are required in mild to moderate renal or hepatic impairment. The use of meloxicam in patients with severe renal or hepatic impairment has not been studied. FDA prescribing information advises against it. The absolute bioavailability of meloxicam capsules was 89% following a single oral dose of 30 mg compared with 30 mg iv bolus injection. Following single intravenous doses, dose-proportional pharmacokinetics were shown in the range of 5 mg to 60 mg. After multiple oral doses the pharmacokinetics of meloxicam capsules were dose-proportional over the range of 7.5 mg to 15 mg. Mean Cmax was achieved within four to five hours after a 7.5 mg meloxicam tablet was taken under fasted conditions, indicating a prolonged drug absorption. With multiple dosing, steady state concentrations were reached by Day 5. A second meloxicam concentration peak occurs around 12 to 14 hours post-dose suggesting biliary recycling. Administration of meloxicam capsules following a high fat breakfast (75 g of fat) resulted in mean peak drug levels (ie, Cmax) being increased by approximately 22% while the extent of absorption (AUC) was unchanged. The time to maximum concentration (Tmax) was achieved between 5 and 6 hours. The mean volume of distribution (Vss) of meloxicam is approximately 10 L. Meloxicam is about 99.4% bound to human plasma proteins (primarily albumin) within the therapeutic dose range. The fraction of protein binding is independent of drug concentration, over the clinically relevant concentration range, but decreases to about 99% in patients with renal disease. Meloxicam penetration into human red blood cells, after oral dosing, is less than 10%. Following a radiolabeled dose, over 90% of the radioactivity detected in the plasma was present as unchanged meloxicam. For more Absorption, Distribution and Excretion (Complete) data for Meloxicam (14 total), please visit the HSDB record page. Metabolism / Metabolites Meloxicam is almost completely metabolized. CYP2C9 is the main enzyme responsible for the metabolism of meloxicam with minor contributions from CYP3A4. Meloxicam has 4 major metabolites with no activity determined. About 60% of the ingested dose is metabolized to 5'-carboxy meloxicam from hepatic cytochrome enzyme oxidation of an intermediate metabolite, 5’-hydroxymethylmeloxicam. Two other metabolites are likely produced via peroxidation. Meloxicam is almost completely metabolized to four pharmacologically inactive metabolites. The major metabolite, 5'-carboxy meloxicam (60% of dose), from P-450 mediated metabolism was formed by oxidation of an intermediate metabolite 5'-hydroxymethyl meloxicam which is also excreted to a lesser extent (9% of dose). In vitro studies indicate that cytochrome P-450 2C9 plays an important role in this metabolic pathway with a minor contribution of the CYP 3A4 isozyme. Patients' peroxidase activity is probably responsible for /two other/ metabolites which account for 16% and 4% of the administered dose, respectively. Meloxicam is extensively metabolized to inactive metabolites in the liver, principally via the cytochrome P-450 (CYP) 2C9 isoenzyme, with minor contribution by CYP3A4. The drug and its metabolites are excreted in urine and feces, and meloxicam undergoes substantial biliary secretion and enterohepatic recirculation. The metabolism of Meloxicam (ME) and the cytochrome(s) P450 (CYPs) involved were analysed by using primary human hepatocytes, human liver microsomes and microsomes from recombinant human B-lymphoblastoid cell lines. While human hepatocytes were capable of converting ME to a 5-hydroxymethyl metabolite (M7) and then to a 5-carboxyderivative (M5), human liver microsomes formed mostly only the 5-hydroxymethylderivative. The kinetics of the formation of M7 by human liver microsomes were biphasic with Km = 13.6 +/- 9.5 and 381 +/- 55.2 uM respectively. The corresponding Vmax were 33.7 +/- 24.2 and 143 +/- 83.9 pmol/min/mg protein respectively. CYP2C9 and, to a much lesser extent, CYP3A4 were found to convert ME to M7. The involvement of 2C9 was demonstrated by inhibition of tolbutamide hydroxylase activity in the presence of ME, inhibition of ME metabolism by sulphaphenazole, correlation between ME metabolism and tolbutamide hydroxylase activity and active metabolism of ME by recombinant 2C9. The involvement of 3A4 was shown by inhibition of ME metabolism by ketoconazole, correlation between ME metabolism and nifedipine oxidase activity and metabolism of ME by recombinant 3A4. Kinetics of the formation of M7 by the individual enzymes resulted in a Km = 9.6 uM and Vmax = 8.4 pmol/min/mg protein for 2C9 and a Km = 475 uM and Vmax = 23 pmol/min/mg protein for 3A4. For more Metabolism/Metabolites (Complete) data for Meloxicam (6 total), please visit the HSDB record page. Meloxicam has known human metabolites that include 5-Hydroxymethyl meloxicam. Biological Half-Life The half-life of meloxicam is approximately 20 hours, which is considerably longer than most other NSAIDS. It can therefore be dosed without the need for slow-release formulations. Meloxicam applied together with [bupivacaine] for postsurgical analgesia had a median half-life of 33-42 hours, depending on dose and application site. The mean elimination half-life (t1/2) ranges from 15 hours to 20 hours. The elimination half-life is constant across dose levels indicating linear metabolism within the therapeutic dose range. ... The twenty volunteers can be classified into extensive metabolizers and poor metabolizers according to pharmacokinetic parameters. The main parameters in the two groups obtained were as follows: T 1/2 were 21 +/- 4 and 38 +/- 9 hr, respectively. ... Absorption: In beagle dogs, oral administration of Meloxicam (0.2 mg/kg) showed slow but complete absorption, with peak plasma concentration (Cmax) of 0.8 ± 0.1 μg/mL reached at 6.2 ± 0.8 hours (Tmax). Absolute oral bioavailability was 89 ± 7% [1] - Half-life: In dogs, the elimination half-life (t₁/₂) of Meloxicam was 24.5 ± 3.2 hours, indicating long-lasting plasma concentrations [1] |
| Toxicity/Toxicokinetics |
Hepatotoxicity Prospective studies found that up to 7% of patients taking meloxicam experienced at least transient serum aminotransferase elevations. These frequently resolved even while continuing the drug and without dose modification. Aminotransferase elevations above 3 fold elevated occurred in 1% of patients. Clinically apparent liver injury with jaundice from meloxicam is rare and only individual case reports have been published. The latency to onset in reported cases was short (1 to 5 weeks) and both cholestatic and hepatocellular patterns of enzyme elevations were described. Immunoallergic features are usually not prominent and autoantibodies are rare, although a single case report of autoimmune hepatitis apparently triggered by meloxicam therapy has been published. Recovery is typically rapid once meloxicam is stopped. Meloxicam is rarely mentioned as an etiologic agent in large case series on drug induced liver injury and acute liver failure. Likelihood score: C (probable rare cause of clinical apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Because no information is available on the use of meloxicam during breastfeeding, other agents may be preferred, especially while nursing a newborn or preterm infant. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Meloxicam is about 99.4% protein bound, primarily to albumin. Interactions When meloxicam is administered with aspirin to healthy volunteers, it tended to increase the AUC (10%) and Cmax (24%) of meloxicam. The clinical significance of this interaction is not known; however, as with other NSAIDs concomitant administration of meloxicam and aspirin is not generally recommended because of the potential for increased adverse effects. Concomitant administration of low-dose aspirin with meloxicam may result in an increased rate of GI ulceration or other complications, compared to use of meloxicam alone. Meloxicam is not a substitute for aspirin for cardiovascular prophylaxis. Pretreatment for four days with cholestyramine significantly increased the clearance of meloxicam by 50%. This resulted in a decrease in t1/2, from 19.2 hours to 12.5 hours, and a 35% reduction in AUC. In a study conducted in healthy subjects, mean pre-dose lithium concentration and AUC were increased by 21% in subjects receiving lithium ... BID with meloxicam ... QD as compared to subjects receiving lithium alone. These effects have been attributed to inhibition of renal prostaglandin synthesis by meloxicam. Patients on lithium treatment should be closely monitored for signs of lithium toxicity when meloxicam is introduced, adjusted, or withdrawn. For more Interactions (Complete) data for Meloxicam (7 total), please visit the HSDB record page. Acute oral toxicity: In male Swiss mice, the oral LD₅₀ of Meloxicam was > 200 mg/kg. No mortality or severe clinical signs (e.g., ataxia, gastrointestinal distress) were observed at doses up to 200 mg/kg [3] - Gastrointestinal safety: In mice treated with Meloxicam (10 mg/kg/day, oral, 7 days), no gastric mucosal erosion or ulceration was observed in HE-stained stomach sections [3] - Plasma protein binding: Meloxicam had high plasma protein binding (99.2 ± 0.3%) in dog plasma (concentration range: 0.1-10 μg/mL) [1] |
| References |
[1]. Effect of structural modification of enol-carboxamide-type nonsteroidal antiinflammatory drugs on COX-2/COX-1 selectivity. J Med Chem. 1997 Mar 14;40(6):980-9. [2]. Meloxicam decreases the migration and invasion of CF41.Mg canine mammary carcinoma cells. Oncol Lett. 2017 Aug;14(2):2198-2206. [3]. Rutin and meloxicam attenuate paw inflammation in mice: Affecting sorbitol dehydrogenase activity. J Biochem Mol Toxicol. 2018 Feb;32(2). |
| Additional Infomation |
Therapeutic Uses Thiazines, Thiazoles; Isoenzymes/antagonists & inhibitors Meloxicam is indicated for relief of the signs and symptoms of osteoarthritis. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals. /Included in US product label/ Meloxicam is used for the management of the signs and symptoms of rheumatoid arthritis in adults. In the management of rheumatoid arthritis in adults, NSAIAs may be useful for initial symptomatic treatment; however, NSAIAs do not alter the course of the disease or prevent joint destruction. /Included in US product label/ Meloxicam is used for the management of the signs and symptoms of pauciarticular or polyarticular course juvenile rheumatoid arthritis in children 2 years of age or older. /NOT included in US product label/ For more Therapeutic Uses (Complete) data for Meloxicam (7 total), please visit the HSDB record page. Drug Warnings /BOXED WARNING/ WARNING: Cardiovascular Risk NSAIDs may cause an increased risk of serious cardiovascular thrombotic events, myocardial infarction, and stroke, which can be fatal. This risk may increase with duration of use. Patients with cardiovascular disease or risk factors for cardiovascular disease may be at greater risk. Meloxicam is contraindicated for the treatment of peri-operative pain in the setting of coronary artery bypass graft (CABG) surgery. /BOXED WARNING/ WARNING: Gastrointestinal Risk: NSAIDs cause an increased risk of serious gastrointestinal adverse events including bleeding, ulceration, and perforation of the stomach or intestines, which can be fatal. These events can occur at any time during use and without warning symptoms. Elderly patients are at greater risk for serious gastrointestinal events. Contraindications: Known hypersensitivity to meloxicam or any ingredient in the formulation. History of urticaria, angioedema, bronchospasm, severe rhinitis, or shock precipitated by aspirin or other NSAIAs. History of aspirin triad (aspirin sensitivity, asthma, and nasal polyps). Treatment of perioperative pain in the setting of coronary artery bypass graft (CABG) surgery. Selective COX-2 inhibitors have been associated with an increased risk of serious adverse cardiovascular thrombotic events in certain situations. Several prototypical NSAIAs also have been associated with an increased risk of cardiovascular events. Findings from a recent systematic review of controlled observational studies and a meta-analysis of published and unpublished data from randomized studies of these agents suggest that use of celecoxib (dosage exceeding 200 mg daily), diclofenac, or indomethacin is associated with an increased risk of cardiovascular events. The possibility exists that meloxicam and ibuprofen also are associated with increased cardiovascular risk. For more Drug Warnings (Complete) data for Meloxicam (25 total), please visit the HSDB record page. Pharmacodynamics Meloxicam is an anti-inflammatory, analgesic analgesic with antipyretic effects in fever. Prostaglandins are substances that contribute to inflammation. This drug also exerts preferential actions against COX-2, which may reduce the possible gastrointestinal effects of this drug. In humans, meloxicam has demonstrated the ability to decrease erythrocyte sedimentation rate(ESR) in patients with rheumatoid arthritis, and to decrease ESR, C-reactive protein (CRP), as well as aquaporin-1 expression. As with other NSAIDS, prolonged use of meloxicum can result in renal or cardiovascular impairment or thrombotic cardiovascular events. A note on gastrointestinal effects As meloxicam preferentially inhibits COX-2, it is thought to cause less gastrointestinal irritation compared to other NSAIDS. Despite this, it still carries a risk of gastric inflammation, bleeding and ulceration. In one study, patients on meloxicam suffered from gastrointestinal symptoms at a rate of 13% compared to 19% of those on [diclofenac]. GI events were found to be less severe in the meloxicam-treated patients. Meloxicam is a preferential COX-2 inhibitor with high selectivity (COX-1/COX-2 ratio = 450), which reduces the risk of gastrointestinal toxicity compared to non-selective NSAIDs. It is clinically used for rheumatoid arthritis, osteoarthritis, and acute musculoskeletal pain [1] - In canine mammary carcinoma cells, Meloxicam inhibits migration and invasion by downregulating MMP-2 and MMP-9 (key enzymes for extracellular matrix degradation), suggesting potential anti-metastatic activity in veterinary oncology [2] - Meloxicam alleviates inflammation not only via COX-2 inhibition but also by suppressing SDH activity in the polyol pathway, which reduces sorbitol accumulation and oxidative stress in inflamed tissues [3] - The long elimination half-life of Meloxicam (24.5 hours in dogs) allows for once-daily administration, improving patient compliance [1] - Unlike selective COX-2 inhibitors with high cardiovascular risk, Meloxicam has a favorable cardiovascular safety profile at therapeutic doses, though high doses may increase COX-1 inhibition and related side effects [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (7.11 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 2: ≥ 1 mg/mL (2.85 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 10.0 mg/mL clear DMSO stock solution to 400 μL of PEG300 and mix evenly; then add 50 μL of Tween-80 to the above solution and mix evenly; then add 450 μL of normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.8458 mL | 14.2288 mL | 28.4576 mL | |
| 5 mM | 0.5692 mL | 2.8458 mL | 5.6915 mL | |
| 10 mM | 0.2846 mL | 1.4229 mL | 2.8458 mL |