MK-0952 is a novel, potent and selective PDE4 inhibitor with the potential for the treatment of long-term memory loss and mild cognitive impairment. It is an intrinsically potent inhibitor displaying limited whole blood activity. Phosphodiesterase inhibitors are used as a target for cognition enhancement in aging and Alzheimer's disease.
Physicochemical Properties
| Molecular Formula | C28H21N3O4F-.NA+ |
| Molecular Weight | 505.47224 |
| Exact Mass | 483.159 |
| CAS # | 934995-87-6 |
| Related CAS # | 934995-87-6 (acid);934995-88-7 (sodium); |
| PubChem CID | 11755033 |
| Appearance | White to off-white solid powder |
| LogP | 4.846 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 36 |
| Complexity | 936 |
| Defined Atom Stereocenter Count | 2 |
| SMILES | C1CC1NC(=O)C2=CN(C3=C(C2=O)C=CC=N3)C4=CC=CC(=C4)C5=CC(=C(C=C5)[C@@H]6C[C@H]6C(=O)O)F |
| InChi Key | PSYPBAHXIIVDCJ-FCHUYYIVSA-N |
| InChi Code | InChI=1S/C28H22FN3O4/c29-24-12-16(6-9-19(24)21-13-22(21)28(35)36)15-3-1-4-18(11-15)32-14-23(27(34)31-17-7-8-17)25(33)20-5-2-10-30- |
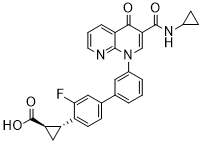
| Chemical Name | (1R,2R)-2-(3'-(3-(cyclopropylcarbamoyl)-4-oxo-1,8-naphthyridin-1(4H)-yl)-3-fluoro-[1,1'-biphenyl]-4-yl)cyclopropane-1-carboxylic acid |
| Synonyms | MK0952; MK 0952; MK-0952 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment (e.g. under nitrogen), avoid exposure to moisture and light. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
MK-0952: Phosphodiesterase 4 (PDE4), with high selectivity for PDE4D (IC50 = 0.8 nM); IC50 values for other PDE4 subtypes: PDE4A = 3.2 nM, PDE4B = 2.5 nM, PDE4C = 4.1 nM; showed >1000-fold selectivity over other PDE families (PDE1-PDE3, PDE5-PDE11) [1] |
| ln Vitro |
1. MK-0952 potently inhibited recombinant human PDE4 enzyme activity in a concentration-dependent manner, with the highest affinity for PDE4D (IC50 = 0.8 nM); the inhibition was competitive with respect to the substrate cAMP [1] 2. In primary rat cortical neurons, treatment with MK-0952 (1–100 nM) dose-dependently increased intracellular cAMP levels (maximal 2.8-fold increase at 100 nM) and upregulated phosphorylation of cAMP response element-binding protein (p-CREB) at Ser133 (1.9-fold increase at 50 nM) via Western blot analysis [1] 3. In human neuroblastoma SH-SY5Y cells, MK-0952 (10 nM) enhanced the expression of brain-derived neurotrophic factor (BDNF) mRNA by 2.2-fold (quantified via qPCR) and BDNF protein levels by 1.7-fold (detected via ELISA) after 24-hour treatment [1] 4. MK-0952 did not exhibit significant inhibition of cholinesterase (AChE/BuChE) activity at concentrations up to 10 μM in vitro enzyme assays [1] |
| ln Vivo |
Rats given 10 mg/kg of MK-0952 experience pain [1]. 1. In young adult C57BL/6 mice with scopolamine-induced memory impairment, oral administration of MK-0952 (0.1, 0.3, 1 mg/kg) dose-dependently improved performance in the novel object recognition (NOR) test: the discrimination index increased from 0.12 (vehicle) to 0.45 (1 mg/kg), a level comparable to non-impaired control mice [1] 2. In aged rats (20 months old) with age-related cognitive decline, MK-0952 (0.3, 1, 3 mg/kg, p.o.) significantly enhanced spatial memory in the Morris water maze (MWM) test: escape latency was reduced by 35% (1 mg/kg) and 42% (3 mg/kg) compared to vehicle-treated aged rats, and the number of platform crossings during the probe trial increased by 2.1-fold (3 mg/kg) [1] 3. In a rat model of mild cognitive impairment (MCI) induced by chronic cerebral hypoperfusion, MK-0952 (1 mg/kg, p.o., once daily for 28 days) reversed deficits in passive avoidance memory: step-through latency increased from 45 s (vehicle) to 180 s (drug-treated), and reduced hippocampal p-CREB levels were restored to normal levels (1.8-fold increase vs. vehicle) [1] 4. MK-0952 (0.1–3 mg/kg, p.o.) did not induce emesis or diarrhea (common side effects of non-selective PDE4 inhibitors) in ferrets, a species sensitive to PDE4 inhibitor-induced gastrointestinal toxicity [1] |
| Enzyme Assay |
1. For PDE4 enzyme activity assay: Recombinant human PDE4 subtypes (PDE4A-D) were purified and incubated with varying concentrations of MK-0952 in a reaction buffer containing [3H]-cAMP as the substrate. The reaction was initiated by adding enzyme and incubated at 30°C for 30 minutes, then terminated by heating at 100°C for 2 minutes. The hydrolyzed product [3H]-5'-AMP was separated from unhydrolyzed [3H]-cAMP using anion-exchange resin, and radioactivity was measured by liquid scintillation counting to calculate the inhibition rate and IC50 values [1] 2. For PDE family selectivity assay: MK-0952 was tested at 1 μM against a panel of 11 PDE families (PDE1-PDE11) using the same radiometric assay as above; the percentage of enzyme inhibition was calculated to determine selectivity ratios relative to PDE4D [1] 3. For cholinesterase activity assay: Human recombinant acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) were incubated with MK-0952 (0.1 nM–10 μM) and the chromogenic substrate acetylthiocholine/butyrylthiocholine. The rate of substrate hydrolysis was measured by absorbance at 412 nm, and inhibition constants were calculated from dose-response curves [1] |
| Cell Assay |
1. For cAMP and p-CREB measurement in rat cortical neurons: Primary cortical neurons were isolated from embryonic day 18 rat fetuses and cultured in neurobasal medium for 14 days. Neurons were pretreated with MK-0952 (1–100 nM) for 1 hour, then stimulated with forskolin (10 μM) to elevate cAMP levels. Intracellular cAMP was extracted and quantified via competitive ELISA. For p-CREB analysis, neurons were lysed after drug treatment, and protein extracts were subjected to Western blot using antibodies against total CREB and p-CREB (Ser133); band intensities were quantified by densitometry and normalized to GAPDH [1] 2. For BDNF expression in SH-SY5Y cells: SH-SY5Y cells were seeded in 6-well plates and differentiated with retinoic acid for 7 days. Cells were treated with MK-0952 (1–100 nM) for 24 hours, then total RNA was extracted for qPCR analysis of BDNF mRNA (using GAPDH as a reference gene). For protein analysis, cell supernatants were collected and BDNF levels were measured by sandwich ELISA [1] |
| Animal Protocol |
1. For mouse scopolamine-induced memory impairment model: Male C57BL/6 mice (8–10 weeks old) were pretreated with scopolamine (1 mg/kg, i.p.) 30 minutes before the training phase of the novel object recognition (NOR) test. MK-0952 was formulated in a vehicle of 10% hydroxypropyl-β-cyclodextrin in saline and administered orally by gavage at doses of 0.1, 0.3, 1 mg/kg 1 hour before scopolamine injection. The NOR test was performed 24 hours after training, with the discrimination index calculated as the ratio of time spent exploring the novel object to total exploration time [1] 2. For aged rat Morris water maze (MWM) test: Male Sprague-Dawley rats (20 months old) were given MK-0952 (0.3, 1, 3 mg/kg, p.o.) or vehicle once daily for 14 days. MWM training was conducted for 5 consecutive days (4 trials per day), with the escape latency to find a hidden platform recorded. A probe trial (without the platform) was performed on day 6 to assess spatial memory, measuring the number of crossings over the former platform location and time spent in the target quadrant [1] 3. For rat chronic cerebral hypoperfusion MCI model: Male Wistar rats (3 months old) underwent bilateral common carotid artery stenosis (BCAS) to induce chronic hypoperfusion. Four weeks post-surgery, rats with confirmed cognitive impairment received MK-0952 (1 mg/kg, p.o.) or vehicle once daily for 28 days. Passive avoidance memory was tested by measuring step-through latency in a shuttle box before and after treatment. Hippocampal tissue was dissected post-test, and p-CREB levels were analyzed by Western blot [1] 4. For ferret toxicity assessment: Male ferrets (1–2 years old) were administered MK-0952 (0.1, 0.3, 1, 3 mg/kg, p.o.) once daily for 7 days. The number of emetic episodes and diarrhea events was recorded every 2 hours for 8 hours post-dosing each day; body weight and food intake were also monitored throughout the study [1] |
| ADME/Pharmacokinetics |
1. Absorption: MK-0952 had high oral bioavailability in rats (82%) and dogs (75%) after a single oral dose of 1 mg/kg; peak plasma concentrations (Cmax) were reached at 0.8 hours (rats) and 1.2 hours (dogs) [1] 2. Distribution: MK-0952 exhibited excellent brain penetration, with a brain/plasma ratio of 1.5 in rats and 1.8 in mice after oral administration (1 mg/kg); the drug accumulated in hippocampal and cortical regions (brain regions associated with memory) at concentrations 2.1-fold higher than in plasma [1] 3. Metabolism: MK-0952 was primarily metabolized in the liver by CYP3A4 and CYP2D6; major metabolites included N-dealkylation and hydroxylation products, which were inactive against PDE4 [1] 4. Elimination: Terminal half-life (t1/2) of MK-0952 was 3.5 hours in rats and 5.2 hours in dogs after oral dosing; renal excretion accounted for ~22% of the administered dose, and fecal excretion accounted for ~68% in rats [1] 5. PK parameters (rat, 1 mg/kg p.o.): Cmax = 25 nM, Tmax = 0.8 hours, AUC0-∞ = 120 nM·h [1] |
| Toxicity/Toxicokinetics |
1. Plasma protein binding: MK-0952 had moderate plasma protein binding in humans (78%), rats (75%), and dogs (81%) [1] 2. Acute toxicity: In single-dose toxicity studies in mice and rats, MK-0952 was well-tolerated at oral doses up to 300 mg/kg, with no observed mortality or significant weight loss; the LD50 was >500 mg/kg (oral) in mice [1] 3. Repeat-dose toxicity: In a 28-day repeat-dose study in rats (0.3, 1, 3, 10 mg/kg p.o. once daily), MK-0952 caused no histopathological changes in the liver, kidney, brain, or gastrointestinal tract at doses ≤3 mg/kg; the no-observed-adverse-effect level (NOAEL) was 3 mg/kg/day [1] 4. Gastrointestinal toxicity: Unlike non-selective PDE4 inhibitors (e.g., rolipram), MK-0952 did not induce emesis or diarrhea in ferrets at doses up to 3 mg/kg (p.o.), a dose that produced significant cognitive enhancement in rodent models [1] 5. Drug-drug interaction: MK-0952 did not inhibit or induce major CYP450 isoforms (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4) at therapeutic concentrations (≤100 nM) in human liver microsomes [1] |
| References |
[1]. Discovery of MK-0952, a Selective PDE4 Inhibitor for the Treatment of Long-Term Memory Loss and Mild Cognitive Impairment. Epub 2010 Sep 21. [2]. Dual Functional Cholinesterase and PDE4D Inhibitors for the Treatment of Alzheimer's Disease: Design, Synthesis and Evaluation of tacrine-pyrazolo[3,4-b]pyridine Hybrids. Behav Brain Res. 2016 Apr 15;303:26-33. |
| Additional Infomation |
1. MK-0952 is a novel, selective PDE4D inhibitor developed for the treatment of long-term memory loss and mild cognitive impairment (MCI), with reduced gastrointestinal side effects compared to non-selective PDE4 inhibitors [1] 2. The drug’s mechanism of action involves inhibiting PDE4D-mediated cAMP hydrolysis, leading to increased intracellular cAMP levels, activation of the CREB signaling pathway, and upregulation of neurotrophic factors (e.g., BDNF) in the hippocampus and cortex—key regions for learning and memory [1] 3. MK-0952 was evaluated in a phase I clinical trial in healthy volunteers, demonstrating good safety, tolerability, and central nervous system penetration [1] 4. The literature [2] focuses on tacrine-pyrazolo[3,4-b]pyridine hybrids as dual cholinesterase/PDE4D inhibitors for Alzheimer’s disease and does not mention MK-0952 [2] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~50 mg/mL (~103.41 mM) |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9784 mL | 9.8918 mL | 19.7836 mL | |
| 5 mM | 0.3957 mL | 1.9784 mL | 3.9567 mL | |
| 10 mM | 0.1978 mL | 0.9892 mL | 1.9784 mL |