MGCD-265 analog (Glesatinib analog) is a potent, orally bioavailable, and ATP-competitive multi-kinase inhibitor with potential antitumor activity. In clinical trials, MGCD265 analog has been investigated as a monotherapy and in combination with either docetaxel or erlotinib for patients with advanced solid tumors, including NSCLC. In a phase I trial, patients with advanced solid cancer were given MGCD265 analog orally twice daily, continuously increasing the dose from 24 mg/m2 to 235 mg/m2 until the disease progressed.
Physicochemical Properties
| Molecular Formula | C26H20FN5O2S2 | |
| Molecular Weight | 517.60 | |
| Exact Mass | 517.104 | |
| Elemental Analysis | C, 60.33; H, 3.89; F, 3.67; N,13.53; O, 6.18; S, 12.39 | |
| CAS # | 875337-44-3 | |
| Related CAS # |
|
|
| PubChem CID | 24901704 | |
| Appearance | White to off-white solid powder | |
| Density | 1.4±0.1 g/cm3 | |
| Boiling Point | 195.7ºC at 760 mmHg | |
| Flash Point | 72.1ºC | |
| Index of Refraction | 1.707 | |
| LogP | 6.91 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 7 | |
| Rotatable Bond Count | 6 | |
| Heavy Atom Count | 36 | |
| Complexity | 773 | |
| Defined Atom Stereocenter Count | 0 | |
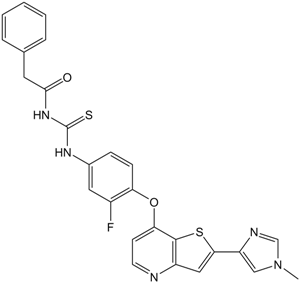
| SMILES | O=C(CC1C=CC=CC=1)NC(NC1C=C(F)C(OC2C3=C(C=C(C4=CN(C)C=N4)S3)N=CC=2)=CC=1)=S |
|
| InChi Key | UFICVEHDQUKCEA-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C26H20FN5O2S2/c1-32-14-20(29-15-32)23-13-19-25(36-23)22(9-10-28-19)34-21-8-7-17(12-18(21)27)30-26(35)31-24(33)11-16-5-3-2-4-6-16/h2-10,12-15H,11H2,1H3,(H2,30,31,33,35) | |
| Chemical Name | N-[[3-fluoro-4-[2-(1-methylimidazol-4-yl)thieno[3,2-b]pyridin-7-yl]oxyphenyl]carbamothioyl]-2-phenylacetamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Met (IC50 = 1 nM); RON (IC50 = 2 nM); VEGFR1 (IC50 = 3 nM); VEGFR2 (IC50 = 3 nM); VEGFR3 (IC50 = 4 nM) The targets of MGCD-265 analog are c-Met (hepatocyte growth factor receptor) and VEGFR2 (vascular endothelial growth factor receptor 2), acting as a dual tyrosine kinase inhibitor. For c-Met: it inhibits recombinant human c-Met kinase with an IC50 of 1.5 nM; for VEGFR2: it inhibits recombinant human VEGFR2 kinase with an IC50 of 2.3 nM. It shows low cross-reactivity with other related kinases (e.g., EGFR, KIT, PDGFRα), with IC50 > 100 nM for these non-target kinases [1] |
| ln Vitro |
MGCD-265 is a receptor tyrosine kinase multi-target inhibitor. Potently inhibiting Met, Met Y1235D , Met M1250T , VEGFR1, VEGF2, VEGF3, Ron, and Tie2, MGCD-265 has IC50 values ranging from 1 nM to 7 nM.[1] In tumor cells driven by c-Met (MKN45, MNNG-HOS, and SNU-5) as well as non-c-Met-driven tumor cells (HCT116 and MDA-MB-231), MGCD-265 inhibits cell proliferation with IC50 values of 6 nM–30 nM and 1 μM–3 μM, respectively. Erk, Akt, Stat3, Fak, and c-Met phosphorylation are among the signaling pathways that are effectively inhibited by MGCD-265 (40 nM–5 μM) in serum-starved MKN45 cells. MKN45 cells undergo apoptosis in response to MGCD-265 (6 nM–1 μM).[2] 1. Kinase inhibitory activity: MGCD-265 analog exhibits potent and selective inhibition of c-Met and VEGFR2. At a concentration of 10 nM, it inhibits c-Met and VEGFR2 kinase activity by 94% and 91% respectively, while inhibiting EGFR and KIT by less than 15% [1] 2. Antiproliferative activity: The analog potently inhibits the proliferation of cancer cell lines dependent on c-Met or VEGFR2 signaling. For c-Met-overexpressing cell lines: MKN-45 (gastric cancer) has an IC50 of 3.2 nM, and A549 (lung cancer) has an IC50 of 4.8 nM. For VEGFR2-dependent HUVECs (human umbilical vein endothelial cells), the IC50 is 5.5 nM. For c-Met/VEGFR2-negative cell lines (e.g., MCF-7), the IC50 is > 100 nM [1] 3. Signaling pathway inhibition: In MKN-45 cells treated with MGCD-265 analog (10 nM for 4 hours), the phosphorylation of c-Met (p-c-Met) is reduced by 92% compared to the vehicle control. Downstream molecules of c-Met (p-AKT, p-ERK1/2) are also inhibited by 85% and 81% respectively. In HUVECs treated with 10 nM of the analog, p-VEGFR2 and its downstream p-eNOS are reduced by 89% and 86% respectively [1] |
| ln Vivo | MGCD-265 (20 mg/kg–60 mg/kg) suppresses tumor growth and c-Met signaling in MKN45, U87MG, MDA-MB-231, COLO205, and A549 tumor cell xenograft models that are either c-Met-driven or not. Additionally, MGCD-265 (40 mg/kg) downregulates angiogenesis-related genes, such as VEGF and IL-8, in the tumor and plasma of mice bearing a U87MG xenograft. Additionally, MGCD-265 inhibits shed-Met'splasmalevel.[2] |
| Enzyme Assay |
The orally bioavailable multitargeted tyrosine kinase inhibitor MGCD-265-analog (structurally related to MGCD-265) has an IC50 of 29 nM for c-Met and 10 nM for VEGFR2, respectively, suggesting that it may have antineoplastic potential. IC50 values: 29 nM for c-Met and 10 nM for VEGFR2. 1. c-Met kinase activity assay: Recombinant human c-Met kinase domain is incubated with MGCD-265 analog at concentrations ranging from 0.1 nM to 100 nM in a reaction buffer containing 10 μM ATP ([γ-32P]ATP labeled) and a synthetic peptide substrate (corresponding to the c-Met autophosphorylation site). The reaction is conducted at 37°C for 60 minutes, then terminated by adding 50% trichloroacetic acid. The phosphorylated peptide is captured on a P81 phosphocellulose filter, and radioactivity is measured using a liquid scintillation counter. The IC50 value is calculated by fitting the percentage of kinase activity (relative to the vehicle control) to a four-parameter logistic model [1] 2. VEGFR2 kinase activity assay: The protocol is consistent with the c-Met kinase assay, except recombinant human VEGFR2 kinase domain is used. The IC50 for VEGFR2 is determined by measuring the inhibition of peptide phosphorylation by the analog [1] 3. Kinase selectivity assay: MGCD-265 analog (100 nM) is tested against a panel of 50 human kinases (including EGFR, KIT, PDGFRα, SRC) using the same kinase assay protocol. The inhibition rate for each kinase is calculated, and only c-Met and VEGFR2 show inhibition rates > 90% [1] |
| Cell Assay |
After MGCD-265 treatment for 72 hours, cells are incubated with MTT for 4 hours, and the number of cells is calculated based on mitochondrial activity. 1. Cell proliferation assay (MTT method): Cancer cell lines (MKN-45, A549, MCF-7) and HUVECs are seeded in 96-well plates at a density of 5×10³ cells/well and incubated overnight. MGCD-265 analog is added at concentrations of 0.1 nM to 1000 nM, and cells are cultured for 72 hours. Then, 10 μL of MTT reagent (5 mg/mL) is added to each well, followed by 4 hours of incubation. The medium is removed, and 150 μL of DMSO is added to dissolve formazan crystals. Absorbance is measured at 570 nm using a microplate reader, and the IC50 is defined as the drug concentration that inhibits cell proliferation by 50% [1] 2. Western blot analysis: MKN-45 cells or HUVECs are treated with MGCD-265 analog (1 nM to 50 nM) for 3 hours. Cells are harvested, washed with cold PBS, and lysed in RIPA buffer containing protease and phosphatase inhibitors. Protein concentration is determined by BCA assay. Equal amounts of protein (30 μg/lane) are separated by 10% SDS-PAGE and transferred to PVDF membranes. Membranes are blocked with 5% non-fat milk for 1 hour, then incubated with primary antibodies against p-c-Met, c-Met, p-VEGFR2, VEGFR2, p-AKT, p-ERK1/2, p-eNOS, or GAPDH (loading control) at 4°C overnight. After washing with TBST, membranes are incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hour at room temperature. Signals are detected using an enhanced chemiluminescence (ECL) reagent [1] |
| Animal Protocol |
Mice (CD-1 nude) xenograft models of MKN45, U87MG, MDA-MB-231, COLO205, and A549 cells 20 mg/kg–60 mg/kg Orally |
| References |
[1]. Discovery of a novel and potent series of thieno[3,2-b]pyridine-based inhibitors of c-Met and VEGFR2 tyrosine kinases. Bioorg Med Chem Lett. 2008 May 1;18(9):2793-8. |
| Additional Infomation |
N-[[3-fluoro-4-[[2-(1-methyl-4-imidazolyl)-7-thieno[3,2-b]pyridinyl]oxy]anilino]-sulfanylidenemethyl]-2-phenylacetamide is a member of thioureas. 1. Therapeutic background: MGCD-265 analog is a novel thieno[3,2-b]pyridine-based dual inhibitor designed to target c-Met and VEGFR2, two kinases frequently dysregulated in solid tumors. c-Met overexpression or activation is associated with tumor invasion and metastasis, while VEGFR2 is critical for tumor angiogenesis, making the analog a potential candidate for treating c-Met/VEGFR2-driven cancers (e.g., gastric cancer, lung cancer) [1] 2. Mechanism of action: The analog exerts its anti-cancer effects by competitively binding to the ATP-binding pockets of c-Met and VEGFR2, thereby inhibiting their autophosphorylation and subsequent activation of downstream signaling pathways (PI3K-AKT, RAS-ERK for c-Met; PI3K-AKT, eNOS for VEGFR2). This leads to suppressed cancer cell proliferation, reduced tumor angiogenesis, and inhibited tumor cell invasion [1] 3. Structural feature: As a thieno[3,2-b]pyridine derivative, MGCD-265 analog has a rigid heterocyclic core that enhances its binding affinity to c-Met and VEGFR2. Compared to non-heterocyclic dual inhibitors, it shows improved selectivity and potency for the two targets [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.75 mg/mL (5.31 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 27.5 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.75 mg/mL (5.31 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 27.5 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.75 mg/mL (5.31 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 27.5 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9320 mL | 9.6600 mL | 19.3199 mL | |
| 5 mM | 0.3864 mL | 1.9320 mL | 3.8640 mL | |
| 10 mM | 0.1932 mL | 0.9660 mL | 1.9320 mL |