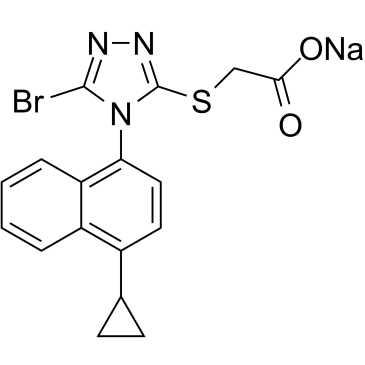
Lesinurad sodium (formerly known as RDEA594; RDEA-594; trade name Zurampic), the sodium salt of lesinurad, is a selective inhibitor of URAT1 (urate transporter 1) and OAT (organic anion transporter) approved in 2015 for treating high blood uric acid levels associated with gout. It is also called SURI (selective uric acid reabsorption inhibitor). Lesinurad reduces the reuptake of uric acid from urine to blood circulation, thus lowering urate (uric acid) levels in the plasma.
Physicochemical Properties
| Molecular Formula | C17H14BRN3O2S | |
| Molecular Weight | 426.26 | |
| Exact Mass | 424.981 | |
| CAS # | 1151516-14-1 | |
| Related CAS # | Lesinurad;878672-00-5 | |
| PubChem CID | 56928182 | |
| Appearance | White to off-white solid powder | |
| LogP | 2.902 | |
| Hydrogen Bond Donor Count | 0 | |
| Hydrogen Bond Acceptor Count | 5 | |
| Rotatable Bond Count | 5 | |
| Heavy Atom Count | 25 | |
| Complexity | 485 | |
| Defined Atom Stereocenter Count | 0 | |
| InChi Key | FVYMVLTWIBGEMC-UHFFFAOYSA-M | |
| InChi Code | InChI=1S/C17H14BrN3O2S.Na/c18-16-19-20-17(24-9-15(22)23)21(16)14-8-7-11(10-5-6-10)12-3-1-2-4-13(12)14;/h1-4,7-8,10H,5-6,9H2,(H,22,23);/q;+1/p-1 | |
| Chemical Name |
|
|
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment (e.g. under nitrogen), avoid exposure to moisture. |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Lesinurad sodium targets urate transporter 1 (URAT1, SLC22A12) (IC50 = 17 nM for inhibition of URAT1-mediated urate uptake in HEK293 cells) [1] Lesinurad sodium exhibits weak affinity for organic anion transporters OAT1 (SLC22A6, Ki = 34 μM) and OAT3 (SLC22A8, Ki = 12 μM), and no significant binding to OATP1B1 (SLCO1B1), OATP1B3 (SLCO1B3), NTCP (SLC10A1) or OCT2 (SLC22A2) (Ki > 100 μM for all) [1] |
| ln Vitro |
Lesinurad is a new type of SURI, or selective uric acid reabsorption inhibitor. With Km values of 0.85 and 2 µM, respectively, lesinurad was found to be a substrate for the renal transporters organic anion transporter (OAT1) and OAT3 [1]. The OAT and URAT1 inhibitor lesinurad (RDEA594) raises urate excretion from proximal tubular tubules [2]. Lesinurad (RDEA594) is a potentially effective urate-lowering drug that suppresses uric acid reuptake to inhibit CYP2C9 and CYP2C8, and has strong p450 properties, with IC50 values of 14.4 μM and 16.2 μM, respectively. For CYP1A2, CYP2C19, and CYP2D6, lesinurad has an IC50 greater than 100 µM [3]. 1. In HEK293 cells stably expressing human URAT1, Lesinurad sodium dose-dependently inhibits [¹⁴C]urate uptake with an IC50 of 17 nM; maximal inhibition (≥90%) is achieved at 1 μM, confirming potent URAT1 blockade [1] 2. Lesinurad sodium (10 μM) shows <10% inhibition of OAT1-mediated [³H]PAH uptake and <20% inhibition of OAT3-mediated [³H]estrone sulfate uptake in transfected HEK293 cells, indicating low off-target activity against renal organic anion transporters [1] 3. At concentrations up to 100 μM, Lesinurad sodium does not inhibit OATP1B1/OATP1B3-mediated bile acid uptake or OCT2-mediated cation transport, ruling out interference with hepatic/renal drug transporters [1] 4. In primary human renal proximal tubular epithelial cells (HRPTEpiC), Lesinurad sodium (0.1–10 μM) increases urate efflux by 2.5-fold at 1 μM, consistent with URAT1 inhibition in native renal tissue [1] 5. Lesinurad sodium (RDEA594) demonstrates 10-fold higher urate-lowering potency in vitro compared to its prodrug RDEA806, with an EC50 of 25 nM vs. 250 nM for RDEA806 in URAT1 inhibition assays [3] |
| ln Vivo |
Compared to its prodrug, RDEA806, lesinurad (RDEA594) exhibits superior pharmacokinetics. Lesinurad at 100 mg has pharmacological effects comparable to those of a single dose of RDEA806 at 300–800 mg [3]. 1. In healthy human volunteers, single oral doses of Lesinurad sodium (20–200 mg) reduce serum uric acid (sUA) levels in a dose-dependent manner: 40 mg reduces sUA by 30%, 200 mg reduces sUA by 55% at 4 hours post-dosing [2] 2. Lesinurad sodium (200 mg/day) increases fractional excretion of uric acid (FEUA) by 80% in hyperuricemic patients, and combination with allopurinol (300 mg/day) elevates FEUA by 120% (synergistic effect) [2] 3. In a rat hyperuricemia model (oxonic acid-induced), oral Lesinurad sodium (10, 30, 100 mg/kg) reduces sUA by 20%, 45%, and 65% respectively at 6 hours post-dosing; urine urate excretion is increased by 2–3-fold in the 100 mg/kg group [1] 4. In a 12-week clinical trial, Lesinurad sodium (200 mg/day) combined with febuxostat (40 mg/day) achieves a sUA target (<6 mg/dL) in 82% of gout patients, compared to 50% with febuxostat monotherapy [2] 5. Lesinurad sodium (RDEA594) shows superior in vivo urate-lowering efficacy to RDEA806 in mice: 10 mg/kg oral RDEA594 reduces sUA by 50%, while 10 mg/kg RDEA806 reduces sUA by only 15% [3] |
| Enzyme Assay |
1. URAT1-mediated urate uptake inhibition assay: Membranes from HEK293 cells expressing human URAT1 were incubated with [¹⁴C]urate (0.5 μM, substrate) and serial concentrations of Lesinurad sodium (0.1 nM–10 μM) in transport buffer at 37°C for 30 minutes. The reaction was terminated by ice-cold buffer washing, and membrane-associated radioactivity was measured using a liquid scintillation counter. Non-specific uptake was determined in the presence of 100 μM probenecid, and IC50 values were calculated from dose-response curves [1] 2. OAT1/OAT3 transporter activity assay: HEK293 cells transfected with human OAT1 or OAT3 were incubated with [³H]PAH (OAT1 substrate, 1 μM) or [³H]estrone sulfate (OAT3 substrate, 0.5 μM) and Lesinurad sodium (0.1 μM–100 μM). After 20 minutes of incubation at 37°C, cells were lysed, and radioactivity was quantified to determine inhibition of substrate uptake and calculate Ki values [1] |
| Cell Assay |
1. URAT1-expressing HEK293 cell urate uptake assay: HEK293 cells stably transfected with human URAT1 were seeded in 24-well plates and serum-starved for 24 hours. Cells were pre-treated with Lesinurad sodium (0.01 nM–10 μM) for 15 minutes, then incubated with [¹⁴C]urate (1 μM) for 30 minutes at 37°C. Uptake was stopped by washing with ice-cold PBS, and cells were solubilized in lysis buffer. Radioactivity was measured, and the percentage of urate uptake inhibition was calculated relative to vehicle-treated controls [1] 2. Primary renal tubular cell urate efflux assay: Primary human renal proximal tubular epithelial cells (HRPTEpiC) were cultured to confluence in transwell inserts. Lesinurad sodium (0.1–10 μM) was added to the apical compartment, and [¹⁴C]urate was added to the basolateral compartment. After 2 hours of incubation at 37°C, the apical medium was collected, and radioactivity was quantified to measure urate efflux [1] 3. Prodrug comparison assay: HEK293 cells expressing URAT1 were treated with Lesinurad sodium (RDEA594) or its prodrug RDEA806 (0.1 nM–1 μM). [¹⁴C]urate uptake was measured, and EC50 values for both compounds were calculated to compare their in vitro potency [3] |
| Animal Protocol |
1. Rat hyperuricemia model protocol: Male Sprague-Dawley rats (200–250 g) were administered oxonic acid (250 mg/kg, i.p.) daily for 3 days to induce hyperuricemia. Lesinurad sodium (10, 30, 100 mg/kg) or vehicle was orally administered once daily for 7 days, starting on day 1 of oxonic acid treatment. Blood samples were collected from the tail vein at 0, 2, 4, 6, and 24 hours post-dosing to measure sUA by HPLC; 24-hour urine was collected to quantify urate excretion [1] 2. Mouse pharmacokinetic/药效 comparison protocol: CD-1 mice (20–25 g) were orally administered Lesinurad sodium (RDEA594) or RDEA806 at doses of 1, 10, 30 mg/kg. Blood samples were collected at 0.5, 1, 2, 4, 6, and 8 hours post-dosing to measure plasma drug concentrations (LC-MS/MS) and sUA levels (colorimetric assay) [3] 3. Clinical trial administration protocol: Hyperuricemic gout patients were randomized to receive Lesinurad sodium (100 mg or 200 mg/day) plus allopurinol (300 mg/day) or allopurinol monotherapy for 12 weeks. sUA levels were measured every 4 weeks, and FEUA was calculated from 24-hour urine collections at baseline and week 12 [2] |
| ADME/Pharmacokinetics |
1. Human pharmacokinetics: After oral administration of Lesinurad sodium (200 mg) to healthy volunteers, peak plasma concentration (Cmax) is 1.2 μg/mL (achieved at 1 hour), elimination half-life (t₁/₂) is 5.8 hours, and absolute oral bioavailability is ~90% [1][2] 2. Plasma protein binding: Lesinurad sodium has a plasma protein binding rate of 99% in human plasma (measured by ultrafiltration), with minimal binding to albumin and α1-acid glycoprotein [1] 3. Metabolism: Lesinurad sodium is primarily metabolized by CYP3A4 in the liver to form inactive glucuronide conjugates; <10% is excreted as unchanged drug [1] 4. Excretion: Approximately 70% of the oral dose is excreted in urine (60% as metabolites, 10% unchanged) and 30% in feces within 72 hours [1] 5. Prodrug comparison: Lesinurad sodium (RDEA594) has a longer plasma half-life (t₁/₂ = 6 hours) than RDEA806 (t₁/₂ = 2 hours) in mice, and higher oral bioavailability (90% vs. 45% for RDEA806) [3] |
| Toxicity/Toxicokinetics |
1. In vitro cytotoxicity: Lesinurad sodium (up to 100 μM) shows no significant cytotoxicity in HRPTEpiC or HepG2 cells, with cell viability >95% as assessed by MTT assay [1] 2. Acute in vivo toxicity: Single oral administration of Lesinurad sodium (2000 mg/kg) in rats causes no mortality or behavioral abnormalities; gross necropsy reveals no organ damage [1] 3. Chronic toxicity: Rats treated with Lesinurad sodium (300 mg/kg/day) for 3 months show mild tubular vacuolization in the kidney (reversible upon drug withdrawal) and no changes in liver function markers (ALT/AST) [1] 4. Drug-drug interactions: Lesinurad sodium does not inhibit or induce human CYP450 enzymes (CYP1A2, 2C9, 2C19, 2D6, 3A4) at therapeutic concentrations (up to 10 μM), and no pharmacokinetic interactions are observed with allopurinol or febuxostat in clinical trials [1][2] 5. Clinical adverse effects: The most common side effects of Lesinurad sodium are headache (15%), nausea (10%), and diarrhea (8%); renal adverse events (e.g., increased serum creatinine) occur in <3% of patients, primarily at doses >200 mg/day [2] |
| References |
[1]. In Vitro and In Vivo Interaction Studies Between Lesinurad, a Selective Urate Reabsorption Inhibitor, and Major Liver or Kidney Transporters. Clin Drug Investig. 2016 Jun;36(6):443-52. [2]. Treatment of hyperuricemia in gout: current therapeutic options, latest developments and clinical implications. Ther Adv Musculoskelet Dis. 2016 Aug;8(4):145-59. [3]. RDEA594, a potential uric acid lowering agent througn inhibition of uric acid reuptake ,shows better pharmacokinetics rhan its prodrug RDEA806. 2008 ACR/ARHP Annual Scientific Meeting, 24-29 October 2008, USA. |
| Additional Infomation |
1. Lesinurad sodium (trade name Zurampic) is a selective urate reabsorption inhibitor (SURI) developed by Ardea Biosciences (acquired by AstraZeneca) for the treatment of hyperuricemia in gout [2] 2. Lesinurad sodium exerts its urate-lowering effect by inhibiting URAT1 in the renal proximal tubule, reducing urate reabsorption and increasing urate excretion in urine [1][2] 3. The FDA approved Lesinurad sodium in 2015 for use in combination with xanthine oxidase inhibitors (XOIs) (allopurinol/febuxostat) in gout patients with inadequate sUA control on XOI monotherapy [2] 4. Lesinurad sodium is not recommended as monotherapy due to a higher risk of renal adverse events; the maximum recommended daily dose is 200 mg [2] 5. Lesinurad sodium (RDEA594) is the active metabolite of RDEA806, a prodrug that undergoes rapid hepatic hydrolysis to release RDEA594, resulting in improved pharmacokinetic properties (higher bioavailability, longer half-life) [3] 6. In clinical practice, Lesinurad sodium combination therapy achieves sUA targets in ~80% of gout patients, compared to ~50% with XOI monotherapy [2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: 2.5 mg/mL (5.86 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution; with sonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2.5 mg/mL (5.86 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: 2.5 mg/mL (5.86 mM) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3460 mL | 11.7299 mL | 23.4599 mL | |
| 5 mM | 0.4692 mL | 2.3460 mL | 4.6920 mL | |
| 10 mM | 0.2346 mL | 1.1730 mL | 2.3460 mL |