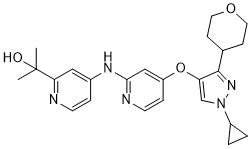
LY3200882 is a next generation, ATP competitive, potent, highly selective small molecule inhibitor of TGF-β receptor type 1 (TGFβRI). It inhibits the serine-threonine kinase domain of TGFβRI. LY3200882 inhibits various pro-tumorigenic activities. LY3200882 potently inhibits TGFβ mediated SMAD phosphorylation in vitro in tumor and immune cells and in vivo in subcutaneous tumors in a dose dependent fashion. In preclinical tumor models, LY3200882 showed potent anti-tumor activity in the orthotopic 4T1-LP model of triple negative breast cancer and this activity correlated with enhanced tumor infiltrating lymphocytes in the tumor microenvironment.
Physicochemical Properties
| Molecular Formula | C24H29N5O3 | |
| Molecular Weight | 435.518765211105 | |
| Exact Mass | 435.227 | |
| Elemental Analysis | C, 66.19; H, 6.71; N, 16.08; O, 11.02 | |
| CAS # | 1898283-02-7 | |
| Related CAS # |
|
|
| PubChem CID | 121249291 | |
| Appearance | Off-white to light yellow solid powder | |
| LogP | 2.1 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 7 | |
| Rotatable Bond Count | 7 | |
| Heavy Atom Count | 32 | |
| Complexity | 612 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | O1CCC(CC1)C1C(=CN(C2CC2)N=1)OC1C=CN=C(C=1)NC1C=CN=C(C=1)C(C)(C)O |
|
| InChi Key | PNPFMWIDAKQFPY-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C24H29N5O3/c1-24(2,30)21-13-17(5-9-25-21)27-22-14-19(6-10-26-22)32-20-15-29(18-3-4-18)28-23(20)16-7-11-31-12-8-16/h5-6,9-10,13-16,18,30H,3-4,7-8,11-12H2,1-2H3,(H,25,26,27) | |
| Chemical Name |
|
|
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
TGF-β receptor type 1/ALK5 (IC50 = 38.2 nM)
Transforming Growth Factor-β type 1 receptor (TGFβRI) (IC50=0.8 nM); exhibits no significant inhibition on TGFβRII (IC50>10000 nM) [2] Transforming Growth Factor-β type 1 receptor (TGFβRI)[1] |
| ln Vitro |
LY3200882 potently suppresses TGFβ induced SMAD phosphorylation in vitro in tumor and immunological cells in a dosage dependent fashion[1]. LY3200882 exhibits potent anti-tumor action in the orthotopic 4T1-LP model of triple negative breast cancer and this activity correlates with augmented tumor infiltrating lymphocytes in the tumor microenvironment[1]. In in vitro immune suppression experiments, LY3200882 has showed the ability to rescue TGFβ1 suppressed or T regulatory cell suppressed naive T cell activity and restore proliferation[1]. LY3200882 decreases NIH3T3 cell viability with an IC50 of 82.9 nM[2]. Mechanism of action studies reveal revealed that LY3200882 inhibits various pro-tumorigenic activities. LY3200882 potently inhibits TGFβ mediated SMAD phosphorylation in vitro in tumor and immune cells. In in vitro immune suppression assays, LY3200882 has shown the ability to rescue TGFβ1 suppressed or T regulatory cell suppressed naïve T cell activity and restore proliferation. Therefore, LY3200882 shows promising activity as an immune modulatory agent. In addition, LY3200882 has shown anti-metastatic activity in vitro in migration assays as well as in vivo in an experimental metastasis tumor model (intravenous EMT6-LM2 model of triple negative breast cancer).[1] Inhibited TGFβRI kinase activity and downstream Smad signaling: LY3200882 potently inhibited recombinant human TGFβRI kinase activity with an IC50 of 0.8 nM, while showing negligible activity against TGFβRII (IC50>10000 nM) and other kinases (selectivity ratio >12500-fold vs TGFβRI). It concentration-dependently blocked TGFβ1-induced Smad2 phosphorylation in A549 cells (IC50=3.2 nM) and MDA-MB-231 cells, as detected by Western blot [2] - Suppressed TGFβ-mediated cell responses: Inhibited TGFβ1-induced epithelial-mesenchymal transition (EMT) in A549 cells, as evidenced by reduced expression of mesenchymal markers (vimentin, N-cadherin) and increased epithelial marker (E-cadherin) via Western blot and immunofluorescence. Also inhibited TGFβ1-induced cell migration and invasion in MDA-MB-231 and A549 cells (p<0.01 vs TGFβ1 alone) [2] - Antiproliferative activity in cancer cells: Exerted concentration-dependent growth inhibition on TGFβ-responsive cancer cell lines, including A549 (IC50=12.5 nM), MDA-MB-231 (IC50=15.8 nM), and HCT116 (IC50=18.3 nM), as measured by MTT assay. Inhibited colony formation of A549 cells with an IC50 of 9.7 nM, showing long-term antiproliferative effects [2] - Blocked TGFβ-induced gene expression: Reduced TGFβ1-induced expression of target genes (PAI-1, CTGF, fibronectin) in A549 cells, as quantified by real-time PCR (p<0.001 vs TGFβ1 alone) [2] - Selective inhibition of TGFβ signaling in vitro: Did not affect BMP (bone morphogenetic protein) signaling or other kinase pathways (e.g., EGFR, VEGFR2, PI3K/Akt) at concentrations up to 10 μM, confirming high target selectivity [1] |
| ln Vivo |
Treatment with LY3200882 (60 mg/kg; oral gavage; twice daily; 21 days; female BALB/C mice) dramatically slows the formation of tumors in the CT26 model[2]. In vivo, LY3200882 significantly and dose-dependently suppresses TGFβ-mediated SMAD phosphorylation in subcutaneous tumors[1]. In an experimental metastasis tumor model (intravenous EMT6-LM2 model of triple negative breast cancer), LY3200882 has demonstrated anti-metastatic efficacy in vivo[1]. In preclinical tumor models, LY3200882 showed potent anti-tumor activity in the orthotopic 4T1-LP model of triple negative breast cancer and this activity correlated with enhanced tumor infiltrating lymphocytes in the tumor microenvironment. Durable tumor regressions in the orthotopic 4T1-LP model were observed and rechallenge of congenic tumors resulted in complete rejection in all mice.Finally, LY3200882 shows combinatorial anti-tumor benefits with checkpoint inhibition (anti-PD-L1) in the syngeneic CT26 model.[1] Inhibited tumor growth in xenograft models: Nude mice bearing subcutaneous A549 or MDA-MB-231 xenografts were treated with LY3200882 (30 mg/kg/day, oral gavage) for 21 days. Tumor volume was measured twice weekly, and final tumor weight was recorded. LY3200882 significantly inhibited tumor growth by 65% (A549) and 58% (MDA-MB-231) compared to vehicle control (p<0.01). Western blot of tumor tissues showed reduced p-Smad2 levels, confirming in vivo inhibition of TGFβRI signaling [1] - Enhanced anti-tumor immune response: In the MC38 syngeneic tumor model, LY3200882 (30 mg/kg/day, oral) combined with anti-PD-1 antibody significantly increased tumor-infiltrating CD8+ T cells and reduced regulatory T cells (Tregs), leading to synergistic tumor growth inhibition (78% inhibition vs vehicle, p<0.001) [1] |
| Enzyme Assay |
ALK5 inhibitory activity[2] A luminescent ADP detection assay was used to assess the ALK5 binding capacity of compounds. Serially dilute the stock solution of 10 mM 3-fold in DMSO to obtain a ten-point dilution curve with final compound concentrations ranging from 3.333 μM to 0.5 nM. Assay measurements were performed in 1X kinase reaction buffer containing 40 mM Tris pH 7.5, 20 mM MgCl2, 0.1% BSA, 1 mM DTT in a final assay volume of 5 μL. Briefly 2.5 μL of ALK5 protein, final concentration of 3 μg/mL, was added to each well of a 384 well assay plate containing 100 nL of each concentration of test compound dissolved in DMSO. 2.5 μL of TGF-βR1 peptide, final concentration 3 μg/mL, and ATP, final concentration 1 mM. Following incubation for 120 min at 28 °C, add 5 μL ADP-Glo™ Reagent to terminate the kinase reaction and deplete the remaining ATP. After incubation for 120 min at 28 °C, add 10 μL of Kinase Detection Reagent to convert ADP to ATP and record luminescence. In vitro p38α enzymatic activity assay[2] All of the enzymatic reactions were conducted at 28 °C for 40 min. The 25 μL reaction mixture contains 50 mM HEPES, pH 7.5, 0.0015% Brij-35, 25 ng kinase, 10 μM ATP, and the FAM-labled peptide. The compounds were tested from 100 μM, 3-fold dilution, 10 concentration. The assay was performed by ChemPartner. It measures kinase activity by quantitating the amount of ATP remaining in solution following a kinase reaction. The luminescent signal from the assay is correlated with the amount of ATP present and is inversely correlated with the amount of kinase activity. The IC50 values were calculated in XLFit excel add-in version 5.4.0.8 to obtain IC50 values using the equation: Y=Bottom + (Top-Bottom)/(1+(IC50/X) ^Hill Slope) where X is Compound Concentration and Y is Inhibition Rate(%). TGFβRI kinase activity assay: Recombinant human TGFβRI kinase domain was incubated with ATP and a specific peptide substrate in the presence of serial dilutions of LY3200882. The phosphorylation of the substrate was detected using a homogeneous time-resolved fluorescence (HTRF) assay. The IC50 value was calculated by plotting the percentage of kinase activity against drug concentration, with vehicle-treated samples set as 100% activity [2] - Kinase selectivity assay: LY3200882 (10 μM) was screened against a panel of 468 human kinases using a radiometric kinase assay. The inhibition rate of each kinase was determined by measuring radioactive ATP incorporation into the substrate, and selectivity was evaluated by comparing inhibition of TGFβRI with other kinases [2] - TGFβRII cross-reactivity assay: Recombinant human TGFβRII kinase domain was used in the same HTRF assay as TGFβRI, with serial concentrations of LY3200882 (0.1 nM–10 μM) to determine IC50 and assess cross-reactivity [2] |
| Cell Assay |
Cell-based luciferase reporter assay for TGF-β type 1 receptor activity[2] The aim of this experiment is to identify compounds which interfere with SMAD 2,3-dependent gene expression selectively in cell-based assays demonstrating that they inhibit ALK5 at cellular level. Plate the Luc-Smad 2/3-NIH3T3 cells from assay-ready frozen stocks at 4000 cells per well in 96-well plates in DMEM medium. After overnight attachment of the cells, media was changed to 2% FBS. Prepare test compounds in DMSO to make 4 mM stock solutions. Serially dilute the stock solutions 4-fold in DMSO to obtain an eight-point dilution curve with final compound concentrations ranging from 20 μM to 1.22 nM and test compounds are added. After 24 h, add Glo Lysis Buffer and Bright-Glo Luciferase assay system to each well to double the well volume. Transfer aliquots (180 μL) to white solid bottom plates for reading luminescence on a plate reader (1 s read). Smad phosphorylation Western blot assay: A549 or MDA-MB-231 cells were serum-starved for 24 hours, pre-treated with LY3200882 (0.1 nM–10 μM) for 1 hour, then stimulated with 5 ng/ml TGFβ1 for 30 minutes. Cells were harvested, proteins extracted, and Western blot was performed with antibodies against p-Smad2, total Smad2, and GAPDH (loading control). Densitometric analysis quantified the ratio of p-Smad2 to total Smad2 [2] - Cell proliferation MTT assay: Cancer cells (A549, MDA-MB-231, HCT116) were seeded in 96-well plates and treated with LY3200882 (0.1 nM–10 μM) for 72 hours. MTT reagent was added, and absorbance was measured after 4 hours of incubation. IC50 values were calculated using dose-response curves [2] - Colony formation assay: A549 cells were seeded in 6-well plates at low density, treated with LY3200882 (0.1 nM–10 μM) for 14 days, with medium changed every 3 days. Colonies were fixed, stained, and counted. The percentage of colony formation was calculated relative to vehicle control [2] - Cell migration and invasion assay: Transwell chambers (with or without Matrigel for invasion) were used. MDA-MB-231 or A549 cells pre-treated with LY3200882 (10 nM–1 μM) for 1 hour were seeded in the upper chamber, and TGFβ1 (5 ng/ml) was added to the lower chamber. After 24 hours (migration) or 48 hours (invasion), cells on the lower membrane were fixed, stained, and counted [2] - EMT marker expression assay: A549 cells were treated with LY3200882 (10 nM–1 μM) and TGFβ1 (5 ng/ml) for 72 hours. Western blot detected E-cadherin, N-cadherin, and vimentin; immunofluorescence staining was used to visualize E-cadherin localization [2] - Target gene expression real-time PCR assay: A549 cells were treated with LY3200882 (0.1 nM–10 μM) and TGFβ1 (5 ng/ml) for 24 hours. Total RNA was isolated, and real-time PCR quantified PAI-1, CTGF, and fibronectin mRNA levels, normalized to GAPDH [2] |
| Animal Protocol |
Animal/Disease Models: BALB/C female mice (5-8 weeks old) injected with CT26 cells[2] Doses: 60 mg/kg Route of Administration: po (oral gavage); twice a day; for 21 days Experimental Results: A statistically significant tumor growth delay in CT26 model was observed. Pharmacokinetics procedures[2] This study was conducted in BALB/C male subjects to investigate the pharmacokinetic profiles of test compounds. The mice were randomized and divided into two groups consisting of 3 mice/group. Prepare test compounds [15r (a LY3200882 analog)] in PEG200-EtOH-solutol-physiological saline (4:1:1:14) to make 0.5 mg/mL solutions for oral gavage and to make 0.1 mg/mL solutions for intravenous injection. Sample collection was performed as follows: 1) single oral administration (PO) group: 5 mg/kg, 0.2 mL/10 g; 2) single tail vein injection (IV) group: 1 mg/kg, 0.1 mL/10 g. Blood was collected from orbital venous plexus in heparinized EP tube at 5, 15, 30 min, 1, 2, 6, 10, 24 h after intravenous or oral administration, and the contents of the blood were analyzed by LC-MS/MS(API 4500). In vivo tumor xenograft model[2] A well-established tumorigenesis assay was used to evaluate the antitumor effect of compound 15r (a LY3200882 analog) in BALB/C female mice model. All mice were housed under standard specific-pathogen-free (SPF) conditions and the animal experiments strictly complied with protocols approved by the Animal Welfare and Ethics Committee (AWEC). 1 × 106 cells/mouse of CT26 cells were injected subcutaneously into the 5-to-8-week-old BALB/C female mice. All compounds were administrated by oral gavage. Mice were examined thrice a week for the development of tumors by palpation, and tumor volumes calculated using formula V = 0.5 × length × width2. The investigators were not blinded to allocation during experiments and outcome assessment. Mice were randomly allocated to three groups consisting of 6 mice/group by an independent person in the laboratory. No statistical method was used to predetermine sample size. The antitumor effect of the compound was assessed by tumor growth inhibition (TGI) or relative tumor proliferation rate (T/C): TGI(%) = [1-(Vt1-Vt0)/(Vc1-Vc0)] × 100%, where Vc1 and Vt1 are the mean volumes of control and treated groups at time of tumor extraction, while Vc0 and Vt0 are the same groups at the start of dosages; T/C (%) = TRTV/CRTV × 100%, where TRTV is the relative tumor volume (RTV) of treated groups, while CRTV is the RTV of control groups. (RTV = Vt/V0, Vt is the mean volumes of treated groups at time of tumor extraction, V0 is the mean volumes of the same groups at the start of dosages). Xenograft tumor growth inhibition assay: Female nude mice (6–8 weeks old) were subcutaneously implanted with 5×10^6 A549 or MDA-MB-231 cells. When tumors reached 100–150 mm³, mice were randomized into vehicle and LY3200882 groups (n=6/group). LY3200882 was administered via oral gavage at 30 mg/kg once daily for 21 days. Tumor volume was measured twice weekly using calipers, and tumor weight was recorded at sacrifice. Tumor tissues were collected for Western blot analysis of p-Smad2 [1] - Syngeneic tumor immunotherapy assay: C57BL/6 mice were subcutaneously implanted with 1×10^6 MC38 colon cancer cells. When tumors reached 80–100 mm³, mice were randomized into four groups: vehicle, LY3200882 (30 mg/kg/day, oral), anti-PD-1 antibody (10 mg/kg, intraperitoneal, twice weekly), or combination. Treatment lasted for 14 days, and tumor volume was measured thrice weekly. Tumor-infiltrating lymphocytes were analyzed by flow cytometry at sacrifice [1] |
| ADME/Pharmacokinetics |
Oral bioavailability: In rats, oral administration of LY3200882 (10 mg/kg) resulted in an oral bioavailability of 52%, with a Cmax of 1.8 μg/ml and AUC0–24h of 12.6 μg·h/ml [1] - Half-life and clearance: In dogs, intravenous administration of LY3200882 (5 mg/kg) showed a terminal half-life (t1/2) of 4.8 hours and total body clearance of 1.2 ml/min/kg. Oral administration (10 mg/kg) yielded a Cmax of 2.3 μg/ml and AUC0–24h of 18.9 μg·h/ml [1] - Plasma protein binding: LY3200882 exhibited high plasma protein binding (>95%) in human, rat, and dog plasma, as determined by equilibrium dialysis [2] |
| Toxicity/Toxicokinetics |
In vitro toxicity: LY3200882 showed no significant cytotoxicity in normal human bronchial epithelial cells (BEAS-2B) at concentrations up to 10 μM (cell viability >85% vs control) [2] - In vivo toxicity: Mice treated with LY3200882 (30 mg/kg/day, oral) for 21 days showed no significant weight loss, hematological abnormalities, or histopathological changes in major organs (liver, kidney, heart, lung) [1] - No drug-drug interaction potential: Did not inhibit cytochrome P450 enzymes (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4) at concentrations up to 10 μM, indicating low potential for drug-drug interactions [2] |
| References |
[1]. Abstract 955: LY3200882, a novel, highly selective TGFβRI small molecule inhibitor. AACR; Cancer Res 2017;77(13 Suppl):Abstract nr 955. [2]. Synthesis and biological evaluation of 4-(pyridin-4-oxy)-3-(3,3-difluorocyclobutyl)-pyrazole derivatives as novel potent transforming growth factor-β type 1 receptor inhibitors. Eur J Med Chem. 2020 Apr 29;198:112354. |
| Additional Infomation |
LY-3200882 is under investigation in clinical trial NCT04158700 (A Study of LY3200882 and Pembrolizumab in Participants With Advanced Cancer). TGFbeta Inhibitor LY3200882 is an orally bioavailable agent that targets transforming growth factor-beta (TGFb), with potential antineoplastic activity. Upon administration, LY3200882 specifically targets and binds to TGFb, which prevents both the binding of TGFb to its receptor TGFbR and TGFb-mediated signal transduction. This may lead to a reduction in TGFb-dependent proliferation of cancer cells. The TGFb signaling pathway is often deregulated in tumors, and plays a key role in the regulation of cell growth, differentiation, apoptosis, motility, invasion, angiogenesis, and various immune responses. The transforming growth factor β (TGFβ) signaling pathway is a pleiotropic cellular pathway that plays a critical role in cancer. In fact, aggressive tumors are typically associated with high ligand levels and thus associated with poor prognosis in various tumor types. Cancer cells use autocrine and paracrine TGFβ signaling to modulate tumor cells and the tumor microenvironment leading to a highly invasive and metastatic phenotype, inducing and increasing tumor vascularization, modulating the extracellular matrix in the stroma, and inhibiting immune surveillance and antitumor immunity. Clinical studies with galunisertib (aka LY2157299 monohydrate), a small molecule inhibitor targeting the TGFβ pathway, have provided proof of concept data supporting the role of TGFβ in cancer and the utility of targeting the TGFβ pathway. Here we describe the identification of LY3200882, a next generation small molecule inhibitor of TGF-β receptor type 1 (TGFβRI). The molecule is a potent, highly selective inhibitor of TGFβRI embodied in a structural platform with a synthetically scalable route. It is an ATP competitive inhibitor of the serine-threonine kinase domain of TGFβRI. In conclusion, we have developed a novel potent and highly selective small molecule inhibitor of TGFβRI for the treatment of cancer.[1] Inhibition of transforming growth factor β (TGF-β) type 1 receptor (ALK5) provides a feasible approach for the treatment of fibrotic diseases and malignant tumors. In this study, we designed and synthesized a new series of 4-(pyridin-4-oxy)-3-(3,3-difluorocyclobutyl)-pyrazole derivatives, and evaluated biologically as TGF-β type 1 receptor inhibitors. The most potent compound 15r inhibited the ALK5 enzyme and NIH3T3 cell viability with IC50 values of 44 and 42.5 nM, respectively. Compound 15r also displayed better oral plasma exposure and excellent bioavailability than LY-3200882, and in vivo inhibited 65.7% of the tumor growth in a CT26 xenograft mouse model.[2] LY3200882 is a novel, highly selective small-molecule inhibitor of TGFβRI, belonging to the 4-(pyridin-4-oxy)-3-(3,3-difluorocyclobutyl)-pyrazole derivative class [2] The mechanism of action involves direct inhibition of TGFβRI kinase activity, thereby blocking the downstream Smad2/3 signaling pathway, which is critical for tumor progression, EMT, metastasis, and immune suppression [1][2] LY3200882 exhibits excellent selectivity for TGFβRI over other kinases (including TGFβRII), minimizing off-target effects [2] In preclinical studies, LY3200882 showed single-agent antitumor activity in TGFβ-responsive xenograft models and synergistic efficacy when combined with immune checkpoint inhibitors (anti-PD-1), highlighting its potential for combination cancer therapy [1] LY3200882 has favorable pharmacokinetic properties, including good oral bioavailability, moderate half-life, and high plasma protein binding, supporting its development as an oral therapeutic agent [1][2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.74 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.74 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.74 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2961 mL | 11.4805 mL | 22.9611 mL | |
| 5 mM | 0.4592 mL | 2.2961 mL | 4.5922 mL | |
| 10 mM | 0.2296 mL | 1.1481 mL | 2.2961 mL |