LDN-212854 (LDN 212854; LDN212854) is a potent and selective inhibitor of BMP (bone morphogenetic protein) receptor with potential antineoplastic activity. It inhibits ALK2 with an IC50 of 1.3 nM. In vivoLDN-212854 potently inhibits heterotopic ossification in an inducible transgenic mutant ALK2 mouse model of fibrodysplasia ossificans progressiva. In vitro, LDN-212854 exhibits some selectivity for ALK2 in preference to other BMP type I receptors, ALK1 and ALK3, which may permit the interrogation of ALK2-mediated signaling, transcriptional activity and function.
Physicochemical Properties
| Molecular Formula | C25H22N6 | |
| Molecular Weight | 406.48 | |
| Exact Mass | 406.19 | |
| Elemental Analysis | C, 73.87; H, 5.46; N, 20.67 | |
| CAS # | 1432597-26-6 | |
| Related CAS # |
|
|
| PubChem CID | 60182388 | |
| Appearance | Light yellow to yellow solid powder | |
| Density | 1.3±0.1 g/cm3 | |
| Index of Refraction | 1.740 | |
| LogP | 1.71 | |
| Hydrogen Bond Donor Count | 1 | |
| Hydrogen Bond Acceptor Count | 5 | |
| Rotatable Bond Count | 3 | |
| Heavy Atom Count | 31 | |
| Complexity | 587 | |
| Defined Atom Stereocenter Count | 0 | |
| InChi Key | BBDGBGOVJPEFBT-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C25H22N6/c1-3-21(22-4-2-10-27-24(22)5-1)23-16-29-31-17-19(15-28-25(23)31)18-6-8-20(9-7-18)30-13-11-26-12-14-30/h1-10,15-17,26H,11-14H2 | |
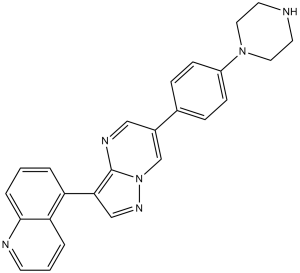
| Chemical Name | 5-(6-(4-(piperazin-1-yl)phenyl)pyrazolo[1,5-a]pyrimidin-3-yl)quinoline | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
ACVR1 (IC50 = 1.3 nM); ALK1 (IC50 = 2.4 nM); BMPR1A (IC50 = 85.8 nM); ALK4 (IC50 = 2133 nM); ALK5 (IC50 = 9276 nM) LDN-212854 is an ALK2-biased inhibitor of bone morphogenetic protein (BMP) type I receptors, targeting ALK2 and ALK3 (ALK2 IC50 = 0.8 nM; ALK3 IC50 = 38 nM) [1] LDN-212854 shows weak inhibition of other ALK receptors (ALK1, ALK4-6: IC50 > 1 μM) and no significant inhibition of unrelated kinases (PKA, PKC: IC50 > 10 μM) [1] |
| ln Vitro |
In BMPR2−/− cells, BMP7-induced phosphorylation of SMAD1/5/8 is blocked by LDN-212854 (0-3.815 μM)[1]. In Huh7 and MT cells, LDN-212854 (2.5 μM, 5 days) suppresses cell proliferation[2]. Huh7 and MT cells' expression of ID1 and EpCAM is suppressed by LDN-212854 (0.5 μM, 48 h)[2]. In recombinant ALK2/ALK3 kinase assays, LDN-212854 dose-dependently inhibits kinase activity. At 10 nM, it inhibits ALK2 activity by 92% and ALK3 activity by 65%, blocking BMP-induced Smad1/5/8 phosphorylation in C2C12 myoblasts. At 1 μM, it downregulates BMP target gene ID1 mRNA by 78% after 24 hours [1] - In human hepatocellular carcinoma (HCC) cells (HepG2, PLC/PRF/5) with activated BMP9-ID1 signaling, LDN-212854 (2 μM) reduces the proportion of EpCAM-positive cancer stem cells (CSCs) by 62% (flow cytometry) after 48 hours. It inhibits cell proliferation by 58-63% (CCK-8 assay) after 72 hours and reduces colony formation efficiency by 65% (colony formation assay) after 14 days. It also downregulates ID1 mRNA by 70% and EpCAM protein by 68% [2] - In normal human hepatocytes (NHHs), LDN-212854 shows no significant toxicity at concentrations up to 20 μM (cell viability > 85% vs. control) [2] |
| ln Vivo | In an inducible transgenic mutant ALK2 mouse model of fibrodysplasia ossificans progressiva, potently suppresses heterotopic ossification when administered intraperitoneally twice daily for four weeks at a dose of 6 mg/kg [1]. By suppressing ID1, LDN-212854 (intraperitoneal injection, 6 mg/kg, twice daily for 4 weeks) inhibits the growth of HCC tumors in HCC xenograft models[2]. |
| Enzyme Assay |
Kinase Assay[1] Purified recombinant ALK1-5 and other kinase proteins, ATP, ATP [γ-32P], and dephosphorylated casein at final concentrations of 2.5nM, 6μM, 0.05 μCi μL−1, and 0.5 mg mL−1 respectively were aliquoted in kinase buffer containing 0.2% bovine serum albumin supplemented with 10mM MnCL2 into 96-well plates, in combination with inhibitor compounds diluted at varying concentrations in kinase buffer (0.01nM to 10 μM) in triplicate. Positive control samples lacking inhibitor compounds, and negative controls lacking recombinant kinase were also measured in triplicate. The mixture was reacted at RT for 45 min., quenched with a final concentration of 2% phosphoric acid. The reaction mixture was transferred to 96-well P81 phosphocellulose filter plates and bound for 5 min. The plates were washed twenty-times with 150 μL of 1% phosphoric acid solution per well by vacuum manifold. Plates were dried at RT for 1 h, sealed, and assayed with Microscint 20 scintillation fluid using a Spectramax L luminometer using the photon counting setting with an integration time of one second per well. Data was normalized to positive controls at 100% enzyme activity with negative controls being subtracted as background. GraphPad was used for graphing and regression analysis by sigmoidal dose-response with variable Hill coefficient. Luciferase Reporter Assay[1] C2C12 Bre-Luc and 293T CAGA-Luc cells were seeded at 20,000 cells in DMEM supplemented with 2% FBS per well in tissue culture treated 96-well plates. The cells were incubated for 1 h (37°C and 10% CO2) and allowed to settle and attach. Compounds of interest or DMSO were diluted in DMEM and added at final compound concentrations of 1 nM to 10 μM. Cells were then incubated for 30 min. Adenovirus expressing constitutively active BMP and TGF-β type I receptors (Ad.caALK1-5) were added to achieve a multiplicity of infection (MOI) of 100. Plates were incubated overnight at 37°C. Cell viability was assayed with an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) colorimetric assay per the manufacturer’s instructions. Media was discarded, and firefly luciferase activity was measured according to manufacturer’s protocol. Light output was measured using a Spectramax L luminometer with an integration time of one second per well. Data was normalized to 100% of incremental BRE-Luc activity due to adenoviruses specifying caALK1, 2, or 3, or the incremental CAGA-Luc activity due to adenoviruses specifying caALK4 or 5. Graphing and regression analysis by sigmoidal dose-response with variable Hill coefficient was performed using GraphPad Prism software. BMP Induced ALP Activity[1] C2C12 myofibroblasts cells were seeded in 96-well plates at 2,000 cells per well in DMEM supplemented with 2% FBS as previously described. Compounds diluted in DMEM and were added at final concentrations ranging from 1nM to 10 μM, in quadruplicate. BMP4 and BMP6 ligands diluted in DMEM were added to final concentrations of 20 ng mL−1. Positive controls were generated by omitting compounds and negative controls were generated by omitting both compounds and ligands. Cells were incubated for 6 days at 37°C and 5% CO2 and subsequently harvested using 1% Triton X-100. Extracts from each well were incubated at RT for 30 min with alkaline phosphatase (ALP) yellow (pNPP) liquid substrate for ELISA and ALP activity was measured by absorbance at 405 nM per the manufacturer’s instructions. Absorbance data was analyzed with positive controls as 100% ALP activity and negative controls being subtracted as background. Graphing and regression analysis by sigmoidal dose-response with variable Hill coefficient was performed using GraphPad Prism. ALK2/ALK3 kinase activity assay: Purified recombinant human ALK2 or ALK3 was incubated with Smad1-derived substrate peptide and LDN-212854 (0.1 nM-100 nM) in assay buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl₂, 1 mM DTT, 0.1 mM ATP) at 30°C for 60 minutes. Phosphorylated substrate was detected by radiolabeled ATP counting, and IC50 values were calculated from dose-response curves [1] - Surface Plasmon Resonance (SPR) binding assay: LDN-212854 (0.01-10 μM) was injected over sensor chips immobilized with ALK2 or ALK3 extracellular domain in running buffer. Binding affinity (KD) was determined by measuring resonance signals, confirming higher binding affinity to ALK2 (KD = 0.5 nM) than ALK3 (KD = 32 nM) [1] |
| Cell Assay |
Western Blot Analysis[1] Cell Types: BMPR2-deficient pulmonary vascular smooth muscle cells Tested Concentrations: 0, 1, 3, 6, 16, 39, 98, 244, 610, 1530, 3815 nM Incubation Duration: Experimental Results: Inhibited the phosphorylation of SMAD1/5/8 induced by BMP7 with an IC50 value of 37 nM. C2C12 cell BMP signaling assay: C2C12 myoblasts were seeded in 6-well plates at 2×10⁵ cells/well and pretreated with LDN-212854 (0.1-5 μM) for 1 hour, then stimulated with BMP4 (10 ng/mL) for 24 hours. Western blot detected p-Smad1/5/8 and total Smad1; qPCR analyzed ID1 mRNA level [1] - HCC cell CSC and proliferation assay: HepG2 and PLC/PRF/5 cells were seeded in 96-well plates (proliferation) or 6-well plates (colony formation/CSC analysis) at 3×10³ cells/well or 2×10⁵ cells/well respectively. Cells were treated with LDN-212854 (0.5-5 μM) for 48-72 hours (proliferation/CSC) or 14 days (colony formation). CCK-8 assay measured proliferation; flow cytometry (EpCAM antibody staining) analyzed CSC proportion; crystal violet staining quantified colonies; qPCR detected ID1 mRNA; Western blot analyzed EpCAM protein [2] |
| Animal Protocol |
Animal/Disease Models: Murine inducible transgenic ALK2Q207D model of heterotopic ossification[1] Doses: 6 mg/kg Route of Administration: intraperitoneal (ip)injection , twice (two times) daily for 4 weeks Experimental Results: Prevented the formation of heterotopic bone and preserved limb range of motion with minimal or no impairment in the majority of mice. Animal/Disease Models: HCC xenografts (Huh7 or MT cell)[1] Doses: 6 mg/kg Route of Administration: intraperitoneal (ip)injection, twice (two times) daily for 10-14 days. Experimental Results: Inhibited tumor growth and demonstrated less spheroid/colony formation ability than PBS-treated tumor cells. |
| References |
[1]. Development of an ALK2-biased BMP type I receptor kinase inhibitor. ACS Chem Biol. 2013;8(6):1291-302. [2]. BMP9-ID1 signaling promotes EpCAM-positive cancer stem cell properties in hepatocellular carcinoma. Mol Oncol. 2021 Aug;15(8):2203-2218. |
| Additional Infomation |
LDN-212854 is a member of the class of quinolines that is quinoline substituted by a 6-[4-(piperazin-1-yl)phenyl]pyrazolo[1,5-a]pyrimidin-3-yl group at position 5. It is a potent ALK inhibitor (IC50 values of 2.4, 1.3, 85.8, 2,133 and 9,276 nM for ALK1, ALK2, ALK3, ALK4 and ALK5, respectively). It has a role as an antineoplastic agent, an angiogenesis inhibitor and an EC 2.7.11.30 (receptor protein serine/threonine kinase) inhibitor. It is a pyrazolopyrimidine, a member of quinolines and a N-arylpiperazine. LDN-212854 is a potent, ALK2-biased small-molecule inhibitor of BMP type I receptors [1] - Its mechanism of action involves competitive binding to the ATP-binding pocket of ALK2 (with higher affinity than ALK3), inhibiting kinase activity and blocking downstream BMP/Smad1/5/8 signaling pathway activation [1] - LDN-212854 exhibits in vitro inhibitory activity against BMP signaling in myoblasts and HCC cells, and suppresses EpCAM-positive cancer stem cell properties in hepatocellular carcinoma [1][2] - It is used as a tool compound to study ALK2-biased BMP signaling in development, cancer, and other BMP-driven biological processes [1][2] - In HCC, it targets BMP9-ID1 signaling axis to inhibit cancer stem cell maintenance and tumorigenic potential [2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: 2.5 mg/mL (6.15 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with sonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2.5 mg/mL (6.15 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4601 mL | 12.3007 mL | 24.6015 mL | |
| 5 mM | 0.4920 mL | 2.4601 mL | 4.9203 mL | |
| 10 mM | 0.2460 mL | 1.2301 mL | 2.4601 mL |