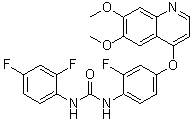
Ki8751 (Ki-8751), a quinolyloxyphenyl-urea analog, is a novel, cell-permeable and selective inhibitor of VEGFR2 (Flk-1) with potential anticancer activity. With an IC50 of 0.9 nM, it inhibits VEGFR2 (Flk-1) and exhibits over 40-fold selectivity for VEGFR2 in comparison to c-Kit (IC50 = 40 nM), PDGFRα (IC50 = 67 nM), and FGFR-2 (IC50 = 170 nM). minimal/none effects on InsR, HGFR, and EGFR. With human tumor xenografts of GL07, St-4, LC6, DLD-1, and A375 cells, it shows strong in vivo antitumor efficacy and outstanding anti-proliferative activity in naked mice and rats.
Physicochemical Properties
| Molecular Formula | C24H18F3N3O4 | |
| Molecular Weight | 469.41 | |
| Exact Mass | 469.124 | |
| Elemental Analysis | C, 61.41; H, 3.87; F, 12.14; N, 8.95; O, 13.63 | |
| CAS # | 228559-41-9 | |
| Related CAS # |
|
|
| PubChem CID | 11317348 | |
| Appearance | White to off-white solid powder | |
| Density | 1.4±0.1 g/cm3 | |
| Boiling Point | 497.1±45.0 °C at 760 mmHg | |
| Melting Point | 239℃ | |
| Flash Point | 254.4±28.7 °C | |
| Vapour Pressure | 0.0±1.3 mmHg at 25°C | |
| Index of Refraction | 1.657 | |
| LogP | 5.91 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 8 | |
| Rotatable Bond Count | 6 | |
| Heavy Atom Count | 34 | |
| Complexity | 677 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | FC1C([H])=C(C([H])=C([H])C=1N([H])C(N([H])C1C([H])=C([H])C(=C([H])C=1F)F)=O)OC1C([H])=C([H])N=C2C([H])=C(C(=C([H])C2=1)OC([H])([H])[H])OC([H])([H])[H] |
|
| InChi Key | LFKQSJNCVRGFCC-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C24H18F3N3O4/c1-32-22-11-15-20(12-23(22)33-2)28-8-7-21(15)34-14-4-6-19(17(27)10-14)30-24(31)29-18-5-3-13(25)9-16(18)26/h3-12H,1-2H3,(H2,29,30,31) | |
| Chemical Name | 1-(2,4-difluorophenyl)-3-[4-(6,7-dimethoxyquinolin-4-yl)oxy-2-fluorophenyl]urea | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.03.00 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
VEGFR2 (IC50 = 0.9 nM); c-Kit (IC50 = 40 nM); PDGFRα (IC50 = 67 nM) The target of Ki8751 is vascular endothelial growth factor receptor 2 (VEGFR-2, also known as KDR). The Ki value for VEGFR-2 is 0.9 nM, and the IC50 value for VEGFR-2 kinase activity inhibition is 1.8 nM. It also shows weak inhibitory activity against other kinases: the IC50 values for VEGFR-1 (Flt-1), PDGFR-β, and c-Kit are 120 nM, 240 nM, and 1100 nM, respectively [1] |
| ln Vitro |
Ki8751 inhibits the phosphorylation of VEGFR-2 at an IC50 value of 0.90 nM. It also blocks the PDGFR family members, including c-Kit and PDGFRR, at 40 nM and 67 nM, respectively. Even at 10000 nM, however, Ki8751 exhibits no inhibitory activity against other kinases, including EGFR, HGFR, InsulinR, and others." At the nanomolar level, VEGF-stimulated human umbilical vein endothelial cells (HUVECs) are inhibited by Ki8751[1]. 1. Inhibition of VEGFR-2-mediated signaling: Treatment with Ki8751 (10 nM) significantly reduces the phosphorylation of VEGFR-2 in human umbilical vein endothelial cells (HUVECs) stimulated by VEGF. It also inhibits the downstream signaling molecules of VEGFR-2, including the phosphorylation of Akt and ERK1/2, with the inhibitory effect being concentration-dependent [1] 2. Antiproliferative activity on endothelial cells: Ki8751 inhibits the proliferation of HUVECs induced by VEGF with an IC50 value of 4.8 nM. It has no significant antiproliferative effect on non-endothelial cells (such as A549 lung cancer cells and MCF-7 breast cancer cells) even at a concentration of 1000 nM [1] 3. Inhibition of endothelial cell migration and tube formation: Ki8751 (1-100 nM) inhibits VEGF-induced HUVEC migration in a concentration-dependent manner. At a concentration of 10 nM, it completely blocks VEGF-induced tube formation of HUVECs on Matrigel [1] |
| ln Vivo |
Ki8751 exhibits noteworthy antitumor activity in nude mice against five human tumor xenografts, including GL07 (glioma), St-4 (stomach carcinoma), LC6 (lung carcinoma), DLD-1 (colon carcinoma), and A375 (melanoma). Additionally, in nude rats, the LC-6 xenograft completely inhibits tumor growth after oral administration once daily for 14 days at a dose of 5 mg/kg without causing any loss of body weight[1]. 1. Antitumor activity in xenograft models: In the A549 human lung cancer xenograft model in nude mice, oral administration of Ki8751 at doses of 10 mg/kg/day and 30 mg/kg/day for 21 days results in tumor growth inhibition rates of 56% and 82%, respectively, compared with the control group. In the MCF-7 human breast cancer xenograft model, oral administration of 30 mg/kg/day of Ki8751 for 21 days achieves a tumor growth inhibition rate of 78% [1] 2. Inhibition of tumor angiogenesis in vivo: Immunohistochemical analysis of tumor tissues from the A549 xenograft model shows that Ki8751 (30 mg/kg/day) reduces the number of CD31-positive blood vessels (a marker of endothelial cells) in tumors by 65% compared with the control group, indicating that it inhibits tumor angiogenesis in vivo [1] |
| Enzyme Assay |
NIH3T3 cells created through human KDR transfection. 1.5 × 104 cells per well are cultured in a 96-well plate coated with collagen type I. Next, a DMEM medium containing 0.1% FCS is added to replace the original medium. After being added to each well, diluted Ki8751 in DMSO is cultured. The cells are stimulated at 37 °C with rhVEGF added at a final concentration of 100 ng/mL. After washing the cells with PBS (pH 7.4), a cell extract is made by adding 50 μL of a solubilization buffer (20 mM HEPES (pH 7.4), 150 mM NaCl, 0.2% Triton X-100, 10% glycerol, 5 mM Na3VO4, 5 mM disodium ethylenediamine tetraacetate, and 2 mM Na4P2O7). ELISA requires the addition of 5 μg/mL of antiphosphotyrosine antibody (PY20) to 50 μL of pH 7.4 PBS to a microplate. Add 300 μL of a blocking solution to the plate after it has been cleaned. Once on the plate, the cell extract is moved. The addition of an anti-VEGFR2 antibody and an anti-rabbit Ig antibody labeled with peroxidase is performed. The absorbance at 450 nm is then measured using a microplate reader after the addition of a chromophoric substrate for peroxidase. The calculation of the VEGFR2 phosphorylation activity for every well involves making the assumption that the absorbance increases to 100% when VEGF is added and to 0% when the test sample is not added. For every instance, the percentage of VEGFR2 phosphorylation that is inhibited is calculated, and the IC50 value is determined. 1. VEGFR-2 kinase activity assay: The assay is conducted in a reaction mixture containing recombinant human VEGFR-2 kinase domain, ATP, and a specific peptide substrate. Ki8751 at different concentrations is added to the reaction mixture, and the mixture is incubated at 30°C for 60 minutes. After the reaction, the amount of phosphorylated peptide substrate is detected using a scintillation proximity assay (SPA). The IC50 value is calculated based on the percentage of kinase activity inhibition at different concentrations of Ki8751 [1] 2. Kinase selectivity assay: The inhibitory activity of Ki8751 against other kinases (VEGFR-1, PDGFR-β, c-Kit, etc.) is determined using the same SPA method as the VEGFR-2 kinase assay. Each kinase reaction is performed with its specific substrate and optimal reaction conditions, and the IC50 values for each kinase are calculated [1] |
| Cell Assay |
HUVECs are plated in type I collagen-precoated 96-well plates at a density of 4000 cells/200 μL/well in order to assess the inhibition of VEGF-Stimulated HUVEC proliferation by Ki8751. Following a 24-hour period, the cells are stimulated with 20 ng/mL rhVEGF after being incubated with Ki8751 for an hour. The cultures are first incubated for 72 hours at 37 °C, followed by a 14-hour re-incubation after receiving a pulse of 1 Ci/well [3H]thymidine. A beta counter is used to measure the incorporation of tritium in cells. 1. HUVEC proliferation assay: HUVECs are seeded in 96-well plates at a density of 2000 cells/well and cultured overnight. Then, VEGF (50 ng/mL) and Ki8751 at different concentrations (0.1-1000 nM) are added to the wells. After culturing for 72 hours, a cell proliferation reagent is added, and the absorbance at 450 nm is measured. The IC50 value for inhibiting VEGF-induced HUVEC proliferation is calculated [1] 2. HUVEC migration assay: A transwell chamber is used. HUVECs are resuspended in medium containing Ki8751 (1-100 nM) and seeded in the upper chamber, while medium containing VEGF (50 ng/mL) is added to the lower chamber. After incubation at 37°C for 6 hours, the cells that migrate to the lower surface of the membrane are fixed, stained, and counted. The migration inhibition rate is calculated based on the number of migrated cells in the treatment group compared with the control group [1] 3. HUVEC tube formation assay: Matrigel is coated on 24-well plates and allowed to solidify. HUVECs are resuspended in medium containing Ki8751 (1-100 nM) and seeded on the Matrigel at a density of 5×104 cells/well. After incubation at 37°C for 18 hours, the tube structures formed by HUVECs are observed under a microscope and quantified by counting the number of tube branches. The inhibition rate of tube formation is calculated [1] 4. Western blot analysis for signaling molecules: HUVECs are serum-starved for 16 hours, then treated with Ki8751 (0.1-100 nM) for 1 hour, followed by stimulation with VEGF (50 ng/mL) for 10 minutes. The cells are lysed, and the lysates are subjected to SDS-PAGE. After transferring to a membrane, the membrane is incubated with primary antibodies against phosphorylated VEGFR-2, phosphorylated Akt, phosphorylated ERK1/2, and total VEGFR-2, Akt, ERK1/2. Then, a secondary antibody conjugated with horseradish peroxidase is added, and the signals are detected using an enhanced chemiluminescence (ECL) system [1] |
| Animal Protocol |
Mice: Human tumor xenografts in nude mice are used to test the effects of Ki8751 on tumor growth against a variety of tumors, including human melanoma (A375), human stomach carcinoma (St-4), human lung carcinoma (LC-6), and human colon carcinoma (DLD-1). For nine days in a row, mice in the experimental groups receive 5 mg/kg of Ki8751 orally once daily, while control animals receive the vehicle. Every two weeks, tumor volumes are checked[1]. 1. Tumor xenograft model establishment: A549 or MCF-7 tumor cells (5×106 cells/mouse) are subcutaneously injected into the right flank of 6-week-old nude mice. When the tumors reach a volume of approximately 100 mm³, the mice are randomly divided into three groups: control group (oral administration of vehicle), low-dose group (oral administration of Ki8751 at 10 mg/kg/day), and high-dose group (oral administration of Ki8751 at 30 mg/kg/day) [1] 2. Drug administration and tumor volume measurement: Ki8751 is dissolved in a vehicle consisting of 0.5% methylcellulose and 0.2% Tween 80. The drug or vehicle is administered orally once a day for 21 consecutive days. Tumor volume is measured every 3 days using a caliper, and the tumor volume is calculated using the formula: Volume = (length × width²)/2 [1] 3. Immunohistochemical analysis of tumor tissues: After the treatment period, the mice are sacrificed, and the tumors are excised. The tumor tissues are fixed in formalin, embedded in paraffin, and cut into 4-μm sections. The sections are incubated with a primary antibody against CD31, followed by a secondary antibody. The sections are then stained with diaminobenzidine (DAB) and counterstained with hematoxylin. The number of CD31-positive blood vessels in five random high-power fields (×400) per section is counted [1] |
| ADME/Pharmacokinetics |
1. Oral bioavailability in mice: After a single oral administration of Ki8751 (30 mg/kg) to mice, the maximum plasma concentration (Cmax) is 1.2 μg/mL, and the area under the plasma concentration-time curve (AUC0-24h) is 8.6 μg·h/mL. After intravenous administration of 10 mg/kg of Ki8751, the AUC0-24h is 3.2 μg·h/mL. The oral bioavailability is calculated to be 88% [1] 2. Plasma half-life in mice: The plasma elimination half-life (t1/2) of Ki8751 in mice after oral administration (30 mg/kg) is 4.2 hours [1] 3. Tissue distribution in mice: At 2 hours after oral administration of 30 mg/kg of Ki8751, the drug concentration in the liver is the highest (8.5 μg/g), followed by the kidney (3.2 μg/g) and tumor tissue (2.1 μg/g). The plasma concentration at this time point is 0.9 μg/mL [1] |
| Toxicity/Toxicokinetics |
1. Acute toxicity in mice: No deaths or obvious toxic symptoms (such as weight loss, lethargy, or abnormal behavior) are observed in mice after a single oral administration of Ki8751 at doses up to 300 mg/kg [1] 2. Subacute toxicity in mice: During the 21-day subacute toxicity study (oral administration of 10 mg/kg/day and 30 mg/kg/day), the body weight of mice in the treatment groups is not significantly different from that of the control group. No obvious pathological changes are found in the major organs (liver, kidney, heart, lung, and spleen) of the mice in the treatment groups through histological examination [1] 3. Plasma protein binding rate: The plasma protein binding rate of Ki8751 in human plasma is 92%, as determined by the equilibrium dialysis method [1] |
| References |
[1]. Novel potent orally active selective VEGFR-2 tyrosine kinase inhibitors: synthesis, structure-activity relationships, and antitumor activities of N-phenyl-N'-{4-(4-quinolyloxy)phenyl}ureas. J Med Chem, 2005, 48(5), 1359-1366. |
| Additional Infomation |
1-(2,4-difluorophenyl)-3-[4-[(6,7-dimethoxy-4-quinolinyl)oxy]-2-fluorophenyl]urea is an aromatic ether. Ki8751 belongs to the class of N-phenyl-N'-{4-(4-quinolyloxy)phenyl}ureas. It is designed as a selective VEGFR-2 inhibitor to target tumor angiogenesis, which is a key process for tumor growth and metastasis. The structure-activity relationship (SAR) study shows that the quinoline ring and urea group in the structure of Ki8751 are crucial for its high affinity and selectivity for VEGFR-2. The introduction of specific substituents on the quinoline ring enhances its inhibitory activity against VEGFR-2 while reducing the inhibitory effect on other kinases [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: 2.5 mg/mL (5.33 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with sonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2.5 mg/mL (5.33 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.33 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 4% DMSO+corn oil: 2.5mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1303 mL | 10.6517 mL | 21.3033 mL | |
| 5 mM | 0.4261 mL | 2.1303 mL | 4.2607 mL | |
| 10 mM | 0.2130 mL | 1.0652 mL | 2.1303 mL |