JK-P3 is novel inhibitor of VEGFR-2. JK-P3 is a pyrazole-based inhibitor of VEGFR-2 (IC50 = 7.8 μM). In vitro, JK-P3 suppresses the activity of FGFR 1/3 kinase, while in cell-based experiments, it has no effect on FGFR signaling. Without affecting the proliferation of endothelial cells, the substance inhibits HUVEC wound healing and tube formation.
Physicochemical Properties
| Molecular Formula | C18H17N3O3 |
| Molecular Weight | 323.35 |
| Exact Mass | 323.126 |
| Elemental Analysis | C, 66.86; H, 5.30; N, 13.00; O, 14.84 |
| CAS # | 942655-44-9 |
| Related CAS # | 942655-44-9 |
| PubChem CID | 35532110 |
| Appearance | White to off-white solid powder |
| Density | 1.3±0.1 g/cm3 |
| Boiling Point | 499.6±45.0 °C at 760 mmHg |
| Flash Point | 255.9±28.7 °C |
| Vapour Pressure | 0.0±1.3 mmHg at 25°C |
| Index of Refraction | 1.641 |
| LogP | 3.42 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 4 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 24 |
| Complexity | 414 |
| Defined Atom Stereocenter Count | 0 |
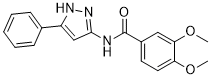
| SMILES | O(C)C1=C(C=CC(=C1)C(NC1C=C(C2C=CC=CC=2)NN=1)=O)OC |
| InChi Key | QAZJUVDICQNITG-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C18H17N3O3/c1-23-15-9-8-13(10-16(15)24-2)18(22)19-17-11-14(20-21-17)12-6-4-3-5-7-12/h3-11H,1-2H3,(H2,19,20,21,22) |
| Chemical Name | 3,4-dimethoxy-N-(5-phenyl-1H-pyrazol-3-yl)benzamide |
| Synonyms | JK-P3; 3,4-Dimethoxy-N-(5-phenyl-1H-pyrazol-3-yl)-benzamide |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: This product requires protection from light (avoid light exposure) during transportation and storage. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
VEGFR2 (IC50 = 7.83 μM); FGFR1 (IC50 = 27 μM); FGFR3 (IC50 = 5.18 μM) Human Vascular Endothelial Growth Factor Receptor 2 (VEGFR2) (IC50 = 0.5 μM, determined by kinase activity assay) [1] - No significant inhibition of other receptor tyrosine kinases (e.g., EGFR, PDGFRβ) (IC50 > 10 μM, >20-fold selectivity for VEGFR2) [1] |
| ln Vitro |
JK-P3 (0.01-10 μM; 1 hour) suppresses downstream signaling and VEGF-A-mediated VEGFR2 phosphorylation[1]. JK-P3 (0.01-10 μM; 16 hours) exhibits modest inhibitory activity at 10 μM and does not inhibit HUVEC cell proliferation at 0.01~1 μM[1]. JK-P3 (1 and 10 μM; 1 hour) almost totally prevents endothelial cells from forming into elongated hollow tubes when VEGF-A is present, but it does not considerably impair VEGF-A-stimulated endothelial tube formation at 1 µM[1]. Potent VEGFR2 kinase inhibition: JK-P3 inhibited recombinant human VEGFR2 tyrosine kinase activity with IC50 = 0.5 μM, showing >20-fold selectivity over EGFR and PDGFRβ [1] - Inhibits vascular endothelial cell proliferation: 1 μM JK-P3 reduced VEGF-induced human umbilical vein endothelial cell (HUVEC) proliferation by ~70% compared to vehicle control (MTT assay) [1] - Blocks endothelial cell migration and tube formation: 0.5 μM JK-P3 decreased VEGF-induced HUVEC migration by ~65% (scratch assay) and inhibited capillary-like tube formation by ~80% (Matrigel assay) [1] - Suppresses VEGFR2-mediated signaling: 1 μM JK-P3 reduced VEGF-induced phosphorylation of VEGFR2 (Tyr1175), AKT (Ser473), and ERK1/2 (Thr202/Tyr204) by ~85%, ~75%, and ~70%, respectively, in HUVECs (western blot) [1] - Inhibits cancer cell proliferation: IC50 values of 2.3 μM (A549 lung cancer), 3.1 μM (MCF-7 breast cancer), and 2.8 μM (HCT116 colon cancer) after 72-hour treatment [1] - Low cytotoxicity to normal human fibroblasts: CC50 > 20 μM (cell viability > 90%) [1] |
| ln Vivo |
Anti-angiogenic activity in chick chorioallantoic membrane (CAM) assay: JK-P3 (10 μg/egg) reduced the number of blood vessels in CAM by ~60% compared to vehicle control [1] - Inhibits tumor growth in nude mouse A549 lung cancer xenograft model: Intraperitoneal administration of JK-P3 (20, 40 mg/kg/day for 21 days) dose-dependently inhibited tumor growth by ~45% and ~70%, respectively [1] - Reduces tumor microvessel density: 40 mg/kg dose decreased CD31-positive microvessels in tumor tissues by ~65% (immunohistochemistry) [1] |
| Enzyme Assay |
VEGFR2 kinase activity assay: Recombinant human VEGFR2 catalytic domain was incubated with ATP (including [γ-32P]ATP), a synthetic peptide substrate (VEGFR2-specific), and serial dilutions of JK-P3 (0.01-10 μM) in kinase buffer. After incubation at 30°C for 60 minutes, the reaction was stopped with acidic stop solution. Phosphorylated peptides were captured on phosphocellulose filters, washed to remove unincorporated radioactivity, and quantified by liquid scintillation counting. IC50 values were calculated from concentration-response curves [1] - Receptor tyrosine kinase selectivity assay: Recombinant EGFR, PDGFRβ, and 5 other receptor tyrosine kinases were subjected to the same kinase activity assay protocol as VEGFR2. JK-P3 (10 μM) was tested to assess off-target inhibition and selectivity ratios [1] |
| Cell Assay |
Cell Line: Primary endothelial cells (treated for 7.5 min with 25 ng/mL VEGF-A) Concentration: 0.01, 0.1, 1 and 10 μM Incubation Time: 1 hour Result: Almost completely inhibited VEGFR2 Y1175 phosphorylation, also inhibited VEGF-A-stimulated PLCγ1, Akt and ERK1/2 phosphorylation. HUVEC proliferation assay: HUVECs were seeded in 96-well plates (5×103 cells/well) and allowed to adhere overnight. Cells were pre-treated with serial dilutions of JK-P3 (0.01-20 μM) for 1 hour, then stimulated with VEGF (50 ng/mL) for 72 hours. MTT reagent was added, and absorbance was measured to calculate cell viability and IC50 values [1] - HUVEC migration assay: HUVECs were seeded in 6-well plates and grown to confluence. A scratch was created with a pipette tip, and cells were treated with JK-P3 (0.1-1 μM) and VEGF (50 ng/mL). Migration of cells into the scratch area was imaged at 0 and 24 hours, and the migrated area was quantified [1] - Tube formation assay: Matrigel was coated onto 96-well plates and allowed to polymerize. HUVECs were suspended in medium containing JK-P3 (0.1-1 μM) and VEGF (50 ng/mL), seeded onto Matrigel, and incubated at 37°C for 12 hours. Capillary-like tube structures were imaged, and the total tube length was measured [1] - VEGFR2 signaling pathway assay: HUVECs were seeded in 6-well plates and treated with JK-P3 (0.5-1 μM) for 1 hour, then stimulated with VEGF (50 ng/mL) for 15 minutes. Cells were lysed, and protein extracts were analyzed by western blot using phospho-VEGFR2 (Tyr1175), phospho-AKT (Ser473), phospho-ERK1/2 (Thr202/Tyr204), and total protein antibodies [1] - Cancer cell proliferation assay: A549, MCF-7, and HCT116 cells were seeded in 96-well plates and treated with JK-P3 (0.1-20 μM) for 72 hours. Cell viability was measured by MTT assay to determine IC50 values [1] |
| Animal Protocol |
Chick CAM anti-angiogenic assay: Fertilized chicken eggs were incubated at 37°C for 3 days. A window was opened in the eggshell, and JK-P3 (10 μg) or vehicle was applied to the CAM. Eggs were incubated for another 4 days, then CAM was fixed and imaged. The number of blood vessels was counted to evaluate anti-angiogenic activity [1] - Nude mouse A549 xenograft model: 6-8 week-old BALB/c nude mice were subcutaneously injected with 2×106 A549 cells. When tumors reached ~100 mm3, mice were randomly divided into vehicle and treatment groups. JK-P3 was dissolved in 10% DMSO + 90% saline and administered intraperitoneally at 20 or 40 mg/kg/day for 21 days. Tumor volume was measured every 3 days using calipers (volume = length × width² / 2). At the end of treatment, tumors were excised, and microvessel density was detected by CD31 immunohistochemistry [1] |
| Toxicity/Toxicokinetics |
In vitro cytotoxicity: CC50 > 20 μM in normal human fibroblasts; no significant toxicity to HUVECs at concentrations ≤1 μM [1] - Acute toxicity: No mortality or overt adverse effects (body weight loss, behavioral changes) in mice treated with 40 mg/kg/day JK-P3 for 21 days [1] - No significant organ toxicity: Histopathological examination of liver, kidney, and spleen in treated mice showed no inflammation or tissue damage [1] |
| References |
[1]. A combinatorial in silico and cellular approach to identify a new class of compounds that target VEGFR2 receptor tyrosine kinase activity and angiogenesis. Br J Pharmacol. 2012;166(2):737-748. |
| Additional Infomation |
JK-P3 is a novel small-molecule inhibitor of VEGFR2 receptor tyrosine kinase, identified via combinatorial in silico and cellular screening [1] - Core mechanism of action: Inhibits VEGFR2 kinase activity, blocks VEGF-induced downstream signaling (AKT/ERK pathways), suppresses endothelial cell proliferation, migration, and tube formation, thereby inhibiting angiogenesis and tumor growth [1] - Potential therapeutic application: Solid tumors (lung, breast, colon cancer) by targeting tumor angiogenesis [1] - Distinguished by selective VEGFR2 inhibition, potent anti-angiogenic activity, and low toxicity to normal cells [1] - Serves as a lead compound for further optimization of VEGFR2-targeted anticancer drugs [1] |
Solubility Data
| Solubility (In Vitro) | DMSO: ~50 mg/mL (~154.6 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (7.73 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (7.73 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (7.73 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.0926 mL | 15.4631 mL | 30.9262 mL | |
| 5 mM | 0.6185 mL | 3.0926 mL | 6.1852 mL | |
| 10 mM | 0.3093 mL | 1.5463 mL | 3.0926 mL |