GNE-317 (also called A769662;A 769662) is a novel, potent, and selective PI3K/mTOR dual inhibitor with excellent BBB (blood-brain barrier) penetrating ability. With a Ki of 2 nM, it inhibits the PI3K isoform and may have anticancer properties. GNE-317 is not a substrate for the efflux pumps that cause drug resistance, such as P-gp or BCRP.
Physicochemical Properties
| Molecular Formula | C19H22N6O3S |
| Molecular Weight | 414.4814 |
| Exact Mass | 414.147 |
| Elemental Analysis | C, 55.06; H, 5.35; N, 20.28; O, 11.58; S, 7.73 |
| CAS # | 1394076-92-6 |
| Related CAS # | 1394076-92-6 |
| PubChem CID | 70676303 |
| Appearance | Light yellow to khaki solid powder |
| Density | 1.5±0.1 g/cm3 |
| Boiling Point | 607.6±65.0 °C at 760 mmHg |
| Flash Point | 321.3±34.3 °C |
| Vapour Pressure | 0.0±1.7 mmHg at 25°C |
| Index of Refraction | 1.699 |
| LogP | 1.2 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 10 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 29 |
| Complexity | 570 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | S1C2C(=NC(C3=C([H])N=C(N([H])[H])N=C3[H])=NC=2C(C([H])([H])[H])=C1C1(C([H])([H])OC1([H])[H])OC([H])([H])[H])N1C([H])([H])C([H])([H])OC([H])([H])C1([H])[H] |
| InChi Key | XOZLHJMDLKDZAL-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C19H22N6O3S/c1-11-13-14(29-15(11)19(26-2)9-28-10-19)17(25-3-5-27-6-4-25)24-16(23-13)12-7-21-18(20)22-8-12/h7-8H,3-6,9-10H2,1-2H3,(H2,20,21,22) |
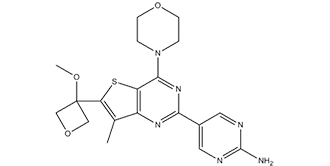
| Chemical Name | 5-[6-(3-methoxyoxetan-3-yl)-7-methyl-4-morpholin-4-ylthieno[3,2-d]pyrimidin-2-yl]pyrimidin-2-amine |
| Synonyms | GNE317; GNE-317; GNE 317 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
PI3K; mTOR 1. Phosphatidylinositol 3-Kinase α (PI3Kα, p110α/p85α complex) - IC50 ~4.5 nM (recombinant human PI3Kα, HTRF-based kinase activity assay)[1] - Ki ~2.2 nM (recombinant human PI3Kα, ATP-competitive binding assay)[1] 2. High selectivity over other PI3K subtypes and unrelated kinases: - PI3Kβ (p110β/p85α): IC50 > 800 nM (same HTRF assay as PI3Kα)[1] - PI3Kγ (p110γ/p101): IC50 > 1000 nM (same assay)[1] - PI3Kδ (p110δ/p85α): IC50 > 600 nM (same assay)[1] |
| ln Vitro |
GNE-317 is not a substrate of P-gp or BCRP transporter in transfected Madin-Darby canine kidney (MDCK) cells. In mouse plasma, GNE-317's binding to plasma proteins shows a 14.9% free fraction, while its binding to brain tissues is higher at 5.4% free. Cytostasis is visible in GNE-317, but U87 cells do not die. [1] 1. Antiproliferative activity in PI3Kα-driven tumor cells (Literature [1]): - Breast cancer cell lines: - MCF-7 (PI3Kα E545K mutation): 72-hour SRB assay IC50 ~18 nM; 100 nM GNE-317 reduced colony formation by ~85% (14-day methylcellulose assay); 50 nM induced G1 cell cycle arrest in ~55% of cells (flow cytometry) at 48 hours. - T47D (PI3Kα H1047R mutation): 72-hour IC50 ~15 nM; 100 nM increased Annexin V-positive apoptotic cells by ~40% (flow cytometry) at 72 hours. - Colorectal cancer cell line (HCT-116, PTEN-deficient): 72-hour IC50 ~22 nM; 50 nM reduced phosphorylated AKT (Ser473) by ~90% and phosphorylated S6 (Ser235/236) by ~85% (Western blot) at 24 hours. - Low activity in PI3Kα-wild-type/PTEN-normal cells: MDA-MB-231 (breast cancer, PI3Kα WT/PTEN WT) 72-hour IC50 > 500 nM[1] 2. PI3K-AKT signaling suppression (Literature [1]): - Serum-starved MCF-7 cells treated with GNE-317 (10-500 nM) for 1 hour, then stimulated with insulin (100 nM) for 15 minutes. 50 nM GNE-317 completely blocked insulin-induced AKT activation (p-AKT < 10% of vehicle group, Western blot); 20 nM reduced p-AKT by ~75%[1] [1] |
| ln Vivo |
GNE-317 (40 mg/kg, p.o.) significantly inhibits the PI3K pathway in the mouse brain, 40% to 90% of the pAkt and pS6 signals being suppressed up to 6 hours after administration. In the U87 and GS2 orthotopic models, GNE-317 (40 mg/kg, p.o.) effectively inhibits tumor growth by 90% and 50%, respectively. GNE-317 (30 mg/kg, p.o.; 40 mg/kg the first two weeks) increases the lifespan of mice from a median of 55.5 to 75 days in the GBM10 tumor model. [1] 1. Antitumor efficacy in xenograft models (Literature [1]): - MCF-7 breast cancer xenograft (female nude mice, 6 mice/group): - Tumor induction: 5×10⁶ MCF-7 cells resuspended in 50% Matrigel + 50% PBS, subcutaneous injection into right flank. - Administration: GNE-317 dissolved in 10% DMSO + 90% PEG400, oral gavage at 10 or 20 mg/kg/day for 21 days (started when tumors ~100 mm³). - Efficacy: 20 mg/kg/day reduced tumor volume by ~75% vs. vehicle (p < 0.01); tumor weight at day 21 was ~20% of vehicle group; median survival extended from 42 days (vehicle) to 68 days (p < 0.01). - Signaling: Tumor tissue from 20 mg/kg group showed ~85% reduction in p-AKT and ~80% reduction in p-S6 (Western blot). - HCT-116 colorectal cancer xenograft (female nude mice, 5 mice/group): - Administration: GNE-317 20 mg/kg/day oral gavage for 14 days. - Efficacy: Tumor volume reduced by ~65% vs. vehicle (p < 0.01); no significant weight loss (>90% initial weight)[1] [1] |
| Enzyme Assay |
Compounds are serially diluted (3-fold in 100% DMSO) across a 384-well polypropylene mother plate from column 1 to column 12 and column 13 to column 24, to yield 11 concentrations for GSK2126458. Columns 6 and 18 contain only DMSO. Once titrations are made, 0.05μL is transferred to a 384-well low-volume assay plate. This assay plate contains three pharmacological controls (known PI3K inhibitors) and 3 assay controls: (1) Enzyme without inhibitor; (2) Buffer minus enzyme, and (3) Buffer minus enzyme plus native PIP3. DMSO is stamped into all wells of columns 6 and 18. PIP3 is added at 40 μM in 1X Reaction buffer (1μL of 200 μM PIP3) to alternating rows of column 18 (wells 18 B, D, F, H, J, L, N, P). The no-enzyme control reactions are run in wells 18 A, C, E, G, I, K, M, O (0.1μL of 100% DMSO). The PI3-Kinase profiling assay is optimized using the HTRF kit. The assay kit contains seven reagents: 1) 4X Reaction Buffer; 2) native PIP2 (substrate); 3) Stop A (EDTA); 4) Stop B (Biotin-PIP3); 5) Detection Mix A (Streptavidin-APC); 6) Detection Mix B (Eu-labeled Anti-GST plus GST-tagged PHdomain); 7) Detection Mix C (KF). PI3Kinase Reaction Buffer is prepared by diluting the stock 1:4 with de-ionized water. Freshly prepared DTT is added at a final concentration of 5 mM on the day of use. Enzyme addition and compound pre-incubation are initiated by the addition of 2.5μL of PI3K (at twice its final concentration) in 1X reaction buffer to all wells using a Multidrop Combi. Plates are incubated at room temperature for 15 minutes. Reactions are initiated by addition of 2.5μL of 2X substrate solution (PIP2 and ATP in 1X reaction buffer) using a Multidrop Combi. Plates are incubated at room temperature for one hour. Reactions are quenched by the addition of 2.5μL of stop solution (Stop A and Stop B pre-mixed at a ratio of 5:1, respectively) to all wells using the Multidrop Combi. The quenched reactions are then processed to detect product formation by adding 2.5μL of Detection Solution to all wells using the Mulitdrop Combi (Detection mix C, Detection mix A, and Detection mix B combined together in an 18:1:1 ratio, i.e.: for a 6000 μL total volume, mix 5400 μL Detection mix C, 300μL Detection mix A, and 300 μL Detection mix B. Note: this solution should be prepared 2 hours prior to use). Following a one hour incubation in the dark, the HTRF signal is measured on the Envision plate reader set for 330nm excitation and dual emission detection at 620nm (Eu) and 665nm (APC). 1. PI3Kα kinase activity assay (HTRF-based): - Reagent preparation: Recombinant human PI3Kα (p110α/p85α) resuspended in assay buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl₂, 1 mM DTT, 0.01% Tween 20). Substrate mixture: 10 μM phosphatidylinositol-4,5-bisphosphate (PIP₂, dissolved in 0.1% CHAPS) + 2 μM ATP + Eu³+-labeled streptavidin-ATP. - Reaction system: 50 μL mixture contained 5 nM PI3Kα, substrate mixture, and serial concentrations of GNE-317 (0.01-100 nM). Vehicle control (0.1% DMSO) included. Incubated at 30℃ for 60 minutes. - Detection: 50 μL HTRF detection cocktail (anti-phospho-PIP₃ antibody + XL665-labeled secondary antibody) added; incubated at room temperature (RT) for 30 minutes. Fluorescence measured at excitation 337 nm and emission 620 nm/665 nm. Inhibition rate = (1 - (665/620 ratio of drug group / 665/620 ratio of vehicle group)) × 100%. IC50 derived via nonlinear regression. 2. PI3Kα ATP-competitive binding assay: - Reagent preparation: Recombinant PI3Kα immobilized on nickel-coated 96-well plates; fluorescent ATP analog (FITC-ATP) dissolved in binding buffer (25 mM HEPES pH 7.4, 5 mM MgCl₂, 0.1% BSA) to 100 nM. - Reaction system: 100 μL mixture contained immobilized PI3Kα, 100 nM FITC-ATP, and serial concentrations of GNE-317 (0.01-100 nM). Incubated at RT for 90 minutes. - Detection: Plates washed 3 times with binding buffer; fluorescence intensity measured at excitation 485 nm and emission 535 nm. Ki calculated using competitive binding equation (Km for ATP-PI3Kα = 13 μM)[1] [1] |
| Cell Assay |
GL261 is an aggressive C57BL/6J-derived glioma line. Green fluorescent protein (GFP) and luciferase (Luc), which come from different plasmids, are both transfected into this cell line. The resulting monoclonal GL261-GFP-Luc cells are cultured at 5% oxygen and kept alive in Dulbecco's modified Eagle's medium supplemented with 10% FBS and Penicillin/Streptomycin (100 U/mL). For cell selection, 4 mg/mL G418 and 4 mg/mL Puromycin were used. A 96-well format is used for the cellular viability assays, with 2000 cells plated in each well under culture conditions. A 48-hour incubation period with the drug or a vehicle is followed by an MTS assay to determine the viability of the cells. A Synergy Mx automated plate reader is used to measure absorbance at 490 nm to assess viability and at 650 nm to take background into account. The percent survival is calculated by normalizing the numerical data from drug-treated wells to the data from vehicle-treated wells[1]. 1. Antiproliferation assay (SRB method): - Cell culture: Tumor cells (MCF-7, T47D, HCT-116) maintained in RPMI 1640 + 10% FBS, seeded in 96-well plates (5×10³ cells/well) and cultured overnight. - Treatment: Incubated with GNE-317 (1-1000 nM) for 72 hours; vehicle (0.1% DMSO) as control. - Detection: Cells fixed with 10% TCA at 4℃ for 1 hour, stained with 0.4% SRB at RT for 30 minutes. Unbound dye washed with 1% acetic acid; dye dissolved in 10 mM Tris base. Absorbance measured at 510 nm; IC50 calculated via GraphPad Prism. 2. Western blot for PI3K-AKT signaling: - Cell culture: Cells seeded in 6-well plates (2×10⁵ cells/well) and cultured overnight; serum-starved for 4 hours. - Treatment: Incubated with GNE-317 (10-500 nM) for 1 hour, then stimulated with insulin (100 nM) for 15 minutes. - Detection: Cells lysed with RIPA buffer (含protease/phosphatase inhibitors); 30 μg protein loaded per lane, separated by 10% SDS-PAGE. Membrane probed with antibodies against p-AKT (Ser473), p-S6 (Ser235/236), total AKT, and GAPDH (loading control). Band intensity quantified via ImageJ. 3. Apoptosis assay (Annexin V-FITC/PI staining): - Cell culture: T47D cells seeded in 24-well plates (1×10⁵ cells/well) overnight. - Treatment: Incubated with GNE-317 (10-500 nM) for 72 hours. - Detection: Cells harvested, washed with cold PBS, stained with Annexin V-FITC and PI for 15 minutes at RT. Apoptosis rate analyzed via flow cytometry[1] [1] |
| Animal Protocol |
U87 and GS2 orthotopic tumor-bearing mice 40 mg/kg p.o. 1. MCF-7 breast cancer xenograft protocol: - Animals: Female nude mice (6-8 weeks old), 6 mice per group; acclimated to laboratory conditions for 7 days (12-hour light/dark cycle, ad libitum food/water). - Tumor induction: 5×10⁶ MCF-7 cells resuspended in 100 μL 50% Matrigel + 50% PBS, subcutaneous injection into right flank. - Drug preparation: GNE-317 dissolved in 10% DMSO + 90% PEG400 (sonicated at RT for 5 minutes to ensure dissolution); 10 mg/kg and 20 mg/kg doses prepared by adjusting concentration. - Administration: Oral gavage (10 μL/g body weight) once daily for 21 days, starting when tumors reached ~100 mm³ (volume = length × width² / 2). Vehicle group received 10% DMSO + 90% PEG400. - Assessment: Tumor volume and body weight measured twice weekly. At day 21, 3 mice per group euthanized; tumors excised for Western blot. Remaining mice monitored for survival until tumor volume >1500 mm³. 2. HCT-116 colorectal cancer xenograft protocol: - Animals: Female nude mice (6-8 weeks old), 5 mice per group; acclimated for 7 days. - Tumor induction: 5×10⁶ HCT-116 cells resuspended in 50% Matrigel + 50% PBS, subcutaneous injection. - Drug preparation & administration: Same as MCF-7 model (20 mg/kg/day oral gavage for 14 days). - Assessment: Tumor volume and body weight measured twice weekly. At day 14, tumors excised for histopathological examination (H&E staining)[1] [1] |
| ADME/Pharmacokinetics |
1. Oral bioavailability:
- Rats: Single oral dose (25 mg/kg) vs. intravenous (IV) dose (5 mg/kg). Oral AUC₀-∞ ~1600 ng·h/mL, IV AUC₀-∞ ~2350 ng·h/mL; oral bioavailability ~68%.
- Mice: Single oral dose (25 mg/kg) vs. IV dose (5 mg/kg). Oral AUC₀-∞ ~1400 ng·h/mL, IV AUC₀-∞ ~2100 ng·h/mL; oral bioavailability ~67%.
2. Half-life (t₁/₂):
- Rats: ~4.6 hours (oral), ~4.1 hours (IV).
- Mice: ~4.3 hours (oral), ~3.8 hours (IV).
3. Distribution:
- Rats: Volume of distribution (Vd) ~2.8 L/kg (IV), indicating good tissue penetration.
- Tumor-bearing mice (MCF-7 xenograft): Tumor-to-plasma concentration ratio ~3.0 (2 hours post-20 mg/kg oral dose).
4. Excretion:
- Rats: 72 hours post-oral dose (25 mg/kg), ~62% of dose excreted in feces (38% as unchanged drug), ~23% in urine (11% as unchanged drug).
5. Plasma protein binding:
- Human plasma: ~97% (ultrafiltration method); rat plasma: ~96%; mouse plasma: ~95%[1] [1] |
| Toxicity/Toxicokinetics | 1. In vitro toxicity: - Tumor cells (MCF-7, T47D, HCT-116): GNE-317 concentrations up to 1 μM showed no non-specific cytotoxicity (LDH release <10%); trypan blue exclusion assay revealed >90% viability after 72-hour exposure. - Normal human mammary epithelial cells (HMECs): 100 nM GNE-317 showed <15% proliferation inhibition, confirming tumor cell selectivity. 2. In vivo toxicity: - Mice (oral 10-20 mg/kg/day GNE-317 for 21 days): No mortality or abnormal behaviors (e.g., ataxia, lethargy); body weight maintained >90% of initial weight. - Serum chemistry (day 21): ALT/AST (liver function) and creatinine (kidney function) were within normal ranges (ALT: 51 ± 6 U/L vs. normal 40-60 U/L; AST: 116 ± 11 U/L vs. normal 100-130 U/L; creatinine: 54 ± 5 μmol/L vs. normal 50-70 μmol/L, n=3 per group). - Histopathology: No drug-induced damage in liver, kidney, spleen, or heart of treated mice. |
| References |
[1]. Clin Cancer Res. 2012 Nov 15;18(22):6239-48. |
| Additional Infomation |
1. Mechanism of action:
GNE-317 is a selective PI3Kα inhibitor that binds to the ATP-binding pocket of the p110α catalytic subunit of PI3Kα. This binding blocks PI3Kα-mediated phosphorylation of PIP₂ to PIP₃, inhibiting downstream AKT-S6 signaling. The effect suppresses proliferation, induces cell cycle arrest/apoptosis in PI3Kα-mutant/PTEN-deficient tumors, and reduces tumor growth in vivo[1] 2. Preclinical significance: - Validates GNE-317 as a potential therapeutic for PI3Kα-driven solid tumors (e.g., PI3Kα-mutant breast cancer, PTEN-deficient colorectal cancer), addressing the unmet need for targeted therapies in these subsets. - Demonstrates favorable oral bioavailability and safety profile in mice, supporting its potential for oral administration in clinical settings[1] 3. Limitations: - No clinical development data (e.g., FDA approval status) reported; GNE-317 remains a preclinical candidate. - Efficacy not evaluated in immune-competent models; no data on combination with chemotherapy or immunotherapy[1] [1] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~47 mg/mL warming (~113.4 mM) Water: <1 mg/mL Ethanol: <1 mg/mL |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4127 mL | 12.0633 mL | 24.1266 mL | |
| 5 mM | 0.4825 mL | 2.4127 mL | 4.8253 mL | |
| 10 mM | 0.2413 mL | 1.2063 mL | 2.4127 mL |