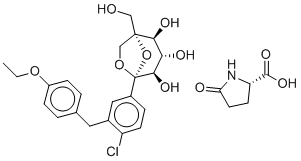
Ertugliflozin L-pyroglutamic acid(formerly known as also known as PF-04971729; trade name: Steglatro), the L-pyroglutamic acid of ertugliflozin, is a potent, orally bioavailable and selective inhibitor of the sodium-dependent glucose cotransporter 2 (SGLT2)approved in 2017 by FDA for treating type 2 diabetes mellitus.
Physicochemical Properties
| Molecular Formula | C27H32CLNO10 | |
| Molecular Weight | 566.0 | |
| Exact Mass | 565.171 | |
| CAS # | 1210344-83-4 | |
| Related CAS # | Ertugliflozin;1210344-57-2 | |
| PubChem CID | 57339449 | |
| Appearance | White to off-white solid powder | |
| Hydrogen Bond Donor Count | 6 | |
| Hydrogen Bond Acceptor Count | 10 | |
| Rotatable Bond Count | 7 | |
| Heavy Atom Count | 39 | |
| Complexity | 740 | |
| Defined Atom Stereocenter Count | 6 | |
| SMILES | CCOC1=CC=C(C=C1)CC2=C(C=CC(=C2)[C@@]34[C@@H]([C@H]([C@@H]([C@@](O3)(CO4)CO)O)O)O)Cl.C1CC(=O)N[C@@H]1C(=O)O |
|
| InChi Key | YHIUPZFKHZTLSH-LXYIGGQGSA-N | |
| InChi Code | InChI=1S/C22H25ClO7.C5H7NO3/c1-2-28-16-6-3-13(4-7-16)9-14-10-15(5-8-17(14)23)22-20(27)18(25)19(26)21(11-24,30-22)12-29-22;7-4-2-1-3(6-4)5(8)9/h3-8,10,18-20,24-27H,2,9,11-12H2,1H3;3H,1-2H2,(H,6,7)(H,8,9)/t18-,19-,20+,21-,22-;3-/m00/s1 | |
| Chemical Name |
|
|
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | In vitro, ertugliflozin L-pyroglutamic acid (PF-04971729 L-pyroglutamic acid) shows a >2000-fold pivoting of SGLT2 inhibition in comparison to SGLT1 [3]. |
| ln Vitro |
In vitro, ertugliflozin L-pyroglutamic acid (PF-04971729 L-pyroglutamic acid) shows a >2000-fold pivoting of SGLT2 inhibition in comparison to SGLT1 [3]. Ertugliflozin demonstrates potent and selective inhibition of human SGLT2 in a functional assay using CHO cells expressing the SGLT transporter, with an IC₅₀ of 0.927 nM. The compound shows >60000-fold selectivity over facilitative glucose transporters (GLUT 1–4). [1] In human hepatocyte (HHEP) incubations, the apparent intrinsic clearance (CLint app) is 4.5 μL/min/million cells, indicating low phase 2 metabolic liability. [1] The compound tests negative in the in vitro micronucleus assay, indicating no clastogenic or aneugenic potential. [1] |
| ln Vivo |
Following shelf release, ertugliflozin L-pyroglutamic acid (PF-04971729 L-pyroglutamic acid) displays concentration-dependent glycosuria [3]. In Sprague–Dawley rats, oral administration of ertugliflozin induces a dose-responsive increase in urinary glucose excretion (UGE) over 24 hours, with maximal UGE of 2554 ± 141.1 mg/200 g body weight at 30 mg/kg. The free in vivo IC₅₀ is 1.8 nM (95% CI = 1.0–2.6 nM). [1] The compound shows robust pharmacodynamic effects in rats, with good agreement between in vitro and in vivo IC₅₀ values for SGLT2 inhibition. [1] |
| Enzyme Assay |
The potency at human SGLT (h-SGLT) was evaluated using a functional assay designed to detect the inhibition of methyl α-D-glucopyranoside (AMG) uptake via the SGLT transporter expressed in Chinese hamster ovary (CHO) cells. The assay measures the inhibition of AMG uptake as a surrogate for glucose transport inhibition. [1] |
| Cell Assay |
The in vitro transport assay for organic anion/cation transporters used human embryonic kidney 293 cells (HEK293) transfected with appropriate transporters (hOAT1, hOAT3, hOCT2). Ertugliflozin showed no uptake by hOAT1, hOAT3, or hOCT2 transporters, indicating low potential for transporter-mediated drug–drug interactions. [1] |
| Animal Protocol |
Male Sprague–Dawley rats (n=5 per group) were randomized to receive one of six doses of ertugliflozin (0.1, 1, 3, 10, 30, and 60 mg/kg) by oral gavage. The formulation used was 20% (v/v) PEG400/24% (v/v) HPBCD. Urine was collected over 24 hours for measurement of UGE. Satellite animals (n=2 per group) were used for drug exposure assessment. [1] For rat intravenous PK studies, male Sprague–Dawley rats (n=2 per group) were administered 2 mg/kg of compound intravenously. The IV formulation for ertugliflozin was DMSO/PEG400/30% SBECD (10/30/60, v/v/v). [1] |
| ADME/Pharmacokinetics |
In Sprague–Dawley rats, following IV administration (2 mg/kg), the total plasma clearance (CLplasma) is 4.04 mL/min/kg, steady-state volume of distribution (Vss) is 1.13 L/kg, and half-life (t₁/₂) is 4.1 hours. [1] Oral administration of the L-pyroglutamic acid cocrystal form (5 mg/kg) in rats resulted in a maximal systemic exposure (Cmax) of 1.94 ± 0.185 μg/mL, time to Cmax of 1.0 hour, and oral bioavailability (F) of 69%. [1] Predicted human pharmacokinetics via allometric scaling: CLplasma = 1.7 mL/min/kg, Vss = 1.8 L/kg, t₁/₂ = 12 hours, and oral bioavailability estimated at 65%. [1] In vitro stability in human liver microsomes (HLM) shows CLint app of 12 μL/min/mg. [1] |
| Toxicity/Toxicokinetics |
Ertugliflozin tested negative in the in vitro mammalian cell micronucleus test, indicating no genotoxic potential under the tested conditions. [1] The compound showed very weak uptake by rat organic cation transporter 2 (rOct2) and organic anion transporter 3 (rOat3), and no uptake by human OAT1, OAT3, or OCT2 transporters, suggesting low risk for transporter-mediated toxicity. [1] |
| References |
[1]. Discovery of a clinical candidate from the structurally unique dioxa-bicyclo[3.2.1]octane class of sodium-dependent glucose cotransporter 2 inhibitors. J Med Chem. 2011 Apr 28;54(8):2952-60. [2]. Pharmacokinetics, metabolism, and excretion of the antidiabetic agent ertugliflozin (PF-04971729) in healthy male subjects. Drug Metab Dispos. 2013 Feb;41(2):445-56. [3]. Preclinical species and human disposition of PF-04971729, a selective inhibitor of the sodium-dependent glucose cotransporter 2 and clinical candidate for the treatment of type 2 diabetes mellitus. Drug Metab Dispos. 2011 Sep;39(9):1609-19. |
| Additional Infomation |
See also: Ertugliflozin (has active moiety); Ertugliflozin pidolate; metformin hydrochloride (component of). Ertugliflozin belongs to a novel class of SGLT2 inhibitors incorporating a dioxa-bicyclo[3.2.1]octane (bridged ketal) ring system, which confers rigidity and may enhance potency and selectivity. [1] The compound is being evaluated in phase 2 clinical trials for the treatment of type 2 diabetes. [1] A pharmacokinetic–pharmacodynamic (PKPD) model was developed to predict human dose and efficacy, which reliably predicted phase 1 clinical trial outcomes. [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (3.67 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (3.67 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (3.67 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.7668 mL | 8.8339 mL | 17.6678 mL | |
| 5 mM | 0.3534 mL | 1.7668 mL | 3.5336 mL | |
| 10 mM | 0.1767 mL | 0.8834 mL | 1.7668 mL |