Physicochemical Properties
| Molecular Formula | C19H14NO4 |
| Molecular Weight | 320.3188 |
| Exact Mass | 320.092 |
| CAS # | 3486-66-6 |
| Related CAS # | Coptisine Sulfate;1198398-71-8;Coptisine chloride;6020-18-4 |
| PubChem CID | 72322 |
| Appearance | Typically exists as solid at room temperature |
| Melting Point | 212-217℃ |
| LogP | -0.87 |
| Hydrogen Bond Donor Count | 0 |
| Hydrogen Bond Acceptor Count | 4 |
| Rotatable Bond Count | 0 |
| Heavy Atom Count | 24 |
| Complexity | 502 |
| Defined Atom Stereocenter Count | 0 |
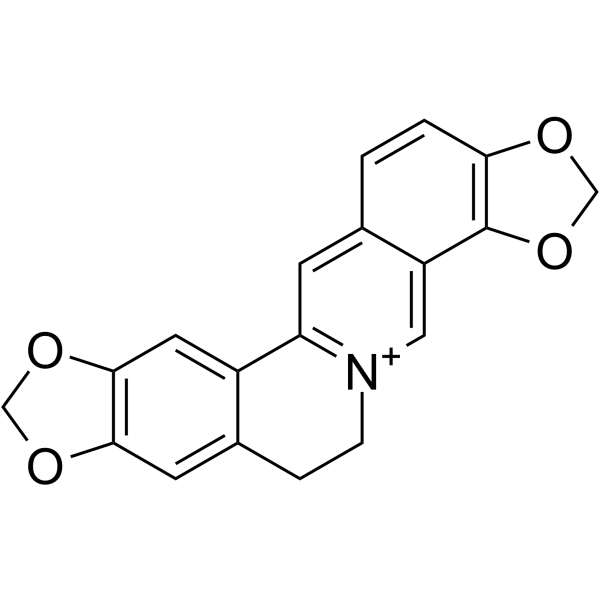
| SMILES | O1C([H])([H])OC2=C1C([H])=C1C(=C2[H])C([H])([H])C([H])([H])[N+]2C([H])=C3C4=C(C([H])=C([H])C3=C([H])C=21)OC([H])([H])O4 |
| InChi Key | XYHOBCMEDLZUMP-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C19H14NO4/c1-2-16-19(24-10-21-16)14-8-20-4-3-12-6-17-18(23-9-22-17)7-13(12)15(20)5-11(1)14/h1-2,5-8H,3-4,9-10H2/q+1 |
| Chemical Name | 5,7,17,19-tetraoxa-13-azoniahexacyclo[11.11.0.02,10.04,8.015,23.016,20]tetracosa-1(13),2,4(8),9,14,16(20),21,23-octaene |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Indoleamine 2,3-dioxygenase (IDO) (Ki = 0.37 μM) [1] |
| ln Vitro |
Coptisine has an IC50 value of 6.3 μM and a Ki value of 5.8 μM, making it a highly effective non-competitive IDO inhibitor[1]. The growth of A549, H460, H2170, MDA-MB-231, and HT-29 cells is inhibited by copoxine (0.1-100 μM), with IC50 values of 18.09, 29.50, 21.60, 20.15, and 26.60 μM, in that order. In A549 cells, coptisine (12.5, 25, and 50 μM) concentration-dependently causes G2/M arrest and apoptosis, downregulates the expression of cyclin B1, cdc2, and cdc25C, and increases the expression of pH2AX and p21. In A549 cells, copoposite (12.5, 25, 50 μM) also causes mitochondrial dysfunction and triggers caspase activity. Additionally, ROS levels are raised by coptisine (50 μM) in a time-dependent manner (0.5, 1, 2, 4, 12, and 24 hours) [3]. - Coptisine (0.1-10 μM) dose-dependently inhibited IDO enzyme activity in IFN-γ-stimulated BV2 microglial cells, reducing the production of kynurenine (KYN) and the KYN/tryptophan (TRP) ratio [1] - Treatment of IFN-γ-stimulated BV2 cells with Coptisine (1, 5, 10 μM) downregulated the mRNA and protein expression of IDO, as detected by qPCR and Western blot [1] - Coptisine (5, 10, 20, 40 μM) inhibited the proliferation of non-small-cell lung cancer A549 cells in a dose- and time-dependent manner (IC50 = 18.7 μM at 48 h, detected by MTT assay) [3] - Coptisine (10, 20, 40 μM) induced G2/M phase cell cycle arrest in A549 cells, as shown by flow cytometry analysis (increased proportion of cells in G2/M phase and decreased proportion in G0/G1 phase) [3] - Coptisine (10, 20, 40 μM) triggered reactive oxygen species (ROS) generation in A549 cells, leading to mitochondria-mediated apoptosis: increased apoptotic rate (Annexin V-FITC/PI staining), decreased mitochondrial membrane potential (JC-1 staining), upregulated Bax/Bcl-2 ratio, and activated caspase-3, -8, -9 (detected by Western blot) [3] - Pretreatment with ROS scavenger N-acetylcysteine (NAC) reversed Coptisine-induced ROS production, mitochondrial dysfunction, and apoptosis in A549 cells [3] |
| ln Vivo |
Mice's LD50 value for coptisine was 880.18 mg/kg, and its toxicity increased with concentration. The dosage of 154 mg/kg/day for 90 days did not cause toxicity in SD rats. In addition to increasing HDL-c content to varied degrees and slowing down the weight gain brought on by the HFHC diet, copoxine (23.35, 46.7, 70.05 mg/kg, po) increased fecal cholesterol and TBA levels in hamsters in a dose-dependent manner. It also reduced TC, TG, and LDL-c levels in the serum of the animals. Inducing the expression of SREBP-2, LDLR, and CYP7A1 proteins involved in cholesterol metabolism, coptisine (70.05 mg/kg, po) lowers the level of HMGCR protein expression [2]. - In APP/PS1 double-transgenic Alzheimer's disease (AD) mice (6-month-old), oral administration of Coptisine (20, 40 mg/kg/day) for 3 months significantly improved cognitive impairment. The mice showed increased escape latency in the Morris water maze test, improved discrimination index in the novel object recognition test, and enhanced step-through latency in the passive avoidance test compared with the vehicle group [1] - Coptisine (20, 40 mg/kg/day) reduced the levels of KYN and KYN/TRP ratio in the serum and brain (hippocampus, cortex) of AD mice, and downregulated IDO mRNA and protein expression in the hippocampus and cortex (detected by qPCR and Western blot) [1] - Coptisine (20, 40 mg/kg/day) decreased the number of amyloid-beta (Aβ) plaques in the hippocampus and cortex of AD mice (detected by immunohistochemistry) [1] - In high-fat diet (HFD)-fed Syrian golden hamsters, oral administration of Coptisine (50, 100 mg/kg/day) for 4 weeks significantly reduced serum total cholesterol (TC), triglycerides (TG), low-density lipoprotein cholesterol (LDL-C), and increased high-density lipoprotein cholesterol (HDL-C) compared with the HFD control group [2] - Coptisine (50, 100 mg/kg/day) decreased hepatic TC and TG levels, and downregulated the mRNA expression of hepatic cholesterol synthesis-related genes (HMGCR, SREBP-2) and lipid accumulation-related gene (PPARγ) in HFD-fed hamsters (detected by qPCR) [2] |
| Enzyme Assay |
- Recombinant human IDO protein was incubated with different concentrations of Coptisine (0.01-10 μM) and L-tryptophan (substrate) in reaction buffer. After incubation at 37°C for 1 hour, the reaction was terminated, and the concentration of KYN (product of IDO-catalyzed reaction) was measured by high-performance liquid chromatography (HPLC) to calculate IDO enzyme activity inhibition rate and Ki value [1] |
| Cell Assay |
- BV2 microglial cell experiment: Cells were seeded and stimulated with IFN-γ (20 ng/mL) for 24 hours to induce IDO expression, then treated with Coptisine (0.1-10 μM) for another 24 hours. qPCR was used to detect IDO mRNA expression (with GAPDH as internal control), Western blot to detect IDO protein level, and HPLC to measure KYN and TRP concentrations in cell supernatants [1] - A549 cell proliferation assay: Cells were seeded in 96-well plates and treated with Coptisine (5-40 μM) for 24, 48, 72 hours. MTT reagent was added, and absorbance at 570 nm was measured to calculate cell viability [3] - A549 cell cycle assay: Cells were treated with Coptisine (10-40 μM) for 24 hours, harvested, fixed with ethanol, stained with propidium iodide (PI), and analyzed by flow cytometry to determine cell cycle distribution [3] - A549 cell apoptosis assay: Cells were treated with Coptisine (10-40 μM) for 24 hours, stained with Annexin V-FITC/PI, and apoptotic rate was detected by flow cytometry; mitochondrial membrane potential was measured by JC-1 staining and flow cytometry [3] - Western blot for A549 cells: Cells were treated with Coptisine (10-40 μM) for 24 hours, lysed to extract proteins, separated by SDS-PAGE, transferred to membranes, and probed with antibodies against Bax, Bcl-2, caspase-3, -8, -9, and β-actin. Bands were visualized and quantified [3] - ROS detection in A549 cells: Cells were loaded with DCFH-DA probe, treated with Coptisine (10-40 μM) for 24 hours (some cells pretreated with NAC for 1 hour), and ROS level was detected by flow cytometry [3] |
| Animal Protocol |
- AD mouse model (APP/PS1 double-transgenic mice): 6-month-old male mice were randomly divided into vehicle group and Coptisine treatment groups (20, 40 mg/kg/day, n=10 per group). Coptisine was dissolved in 0.5% carboxymethylcellulose sodium (CMC-Na) solution. Mice received daily oral gavage for 3 months. Cognitive function tests (Morris water maze, novel object recognition, passive avoidance) were performed before sacrifice. Serum, hippocampus, and cortex tissues were collected for KYN/TRP ratio detection, qPCR, Western blot, and immunohistochemistry [1] - Hypercholesterolemic hamster model: Male Syrian golden hamsters were fed a high-fat diet (HFD) for 2 weeks to induce hypercholesterolemia, then randomly divided into HFD control group and Coptisine treatment groups (50, 100 mg/kg/day, n=8 per group). Normal diet group was set as control. Coptisine was dissolved in distilled water and administered by daily oral gavage for 4 weeks. Body weight was recorded weekly. At the end of treatment, serum was collected to detect lipid profiles (TC, TG, LDL-C, HDL-C), and liver tissue was collected for TC/TG content measurement and qPCR analysis [2] |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Corydalis saxicola Bunting (Yanhuanglian) is an important component in various prescriptions in traditional Chinese medicine. Yanhuanglian has been demonstrated to possess many pharmacological activities, including antibacterial, antiviral, and anticancer activities. The active fractions are dehydrocavidine, coptisine, dehydroapocavidine, and tetradehydroscoulerine. The purpose of the present study was to examine in vivo pharmacokinetics and tissue distribution in rats by using high-performance liquid chromatography (HPLC) coupled with tandem mass spectrometry. Systemic clearance of the four active alkaloids in plasma was over 93% of hepatic blood flow, indicating they may be quickly eliminated via hepatic clearance. Less than 10% drugs was excreted via urine following intravenous and oral administration, suggesting that these four alkaloids may undergo significant metabolism in the body or the drug may be excreted via other routes other than urine. There was significantly lower excretion of these four alkaloids following oral than intravenous administration, suggesting a significant first pass effect after oral administration. There appeared to be wide distribution of those four alkaloids in rats as demonstrated by the higher apparent volume of distribution. Our results have also demonstrated that the four alkaloids can be absorbed following oral administration although there were less than 15% of drugs absorbed into systemic circulation. In summary, the favorable oral bioavailability properties of those four active alkaloids in rats make Yanhuanglian extract worth further investigation for improving oral bioavailability. To study the absorption of coptisine chloride (COP) and berberrubine (BRB) as chemical constituents of some traditional Chinese medicines in human intestinal epithelial. By using Caco-2 (the human colonic adenocarcinoma cell lines) cell monolayers as an intestinal epithelial cell model, the permeability of COP and BRB were studied from apical side (AP side) to basolateral side (BL side) or from BL side to AP side. The two alkaloids were measured by reversed-phase high performance liquid chromatography (HPLC) coupled with UV detector. Transport parameters and apparent permeability coefficients (P(app)) were then calculated and compared with those of propranolol and atenolol. P(app) values were also compared with the reported values for model compounds (propranolol and atenolol). The P(app) values of COP, BRB were (1.103 +/- 0.162) x 10(-5), (1.309 +/- 0.102) x 10(-5) cm x s(-1 from AP side to BL side, and (0.300 +/- 0.041) x 10(-5) and (1.955 +/- 0.055) x 10(-5) cm x s(-1) from BL side to AP side, respectively. Their P(app) values were identical with those of propranolol [(2.23 +/- 0.10) x 10(-5 cm x s(-1)], which is a transcellular transport marker and as a control substance for high permeability. On the other hand, the efflux transport of BRB was higher 1.49 times more than its influx transport with 0.67 rate of P(app A-->B)/P(app B-->A). But P(app A-->B)/P(app B-->A value of COP was 3.67, which suggested that the efflux transport have not been involved in its absorbed mechanism in Caco-2 cells monolayers. COP and BRB can be absorbed across intestinal epithelial cells, and they are completely absorbed compounds. BRB may have been involved in efflux mechanism in Caco-2 cells monolayers model from the basolateral-to-apical direction. To determine the pharmacokinetics, distribution, and mutual transformation of the total alkaloids, jatrorrhizine, coptisine, berberine, and palmatine from Coptis chinensis in rats. After the total alkaloids and berberine were fed into rats, their contents in plasma, tissues and gastrointestinal tract were determined by reversed-phase HPLC. The peak times of berberine in blood were 2.0 hr (Cmax 3.7 mg x L(-1)) and 5.0 hr Cmax 2.8 mg x L(-1)), respectively. Berberine in rat blood can be transformed into jatrorrhizine. After the rats were fed with the total alkaloids by gavage, the content of berberine was decreased monotonously, while coptisine, palmatine, and jatrorrhizine contents were increased gradually in the stomach, it speculated that berberine may be transformed into jatrorrhizine in the stomach. Animal experiments showed that berberine and palmatine were mainly distributed in the lungs of animals, followed by the distribution in the liver, while jatrorrhizine and coptisine was mainly in the liver, then in the lungs. Berberine could transform into jatrorrhizine. The mechanism on the appearance of two maximum blood concentration of berberine in blood could be explained with the propulsion of the gastrointestinal tract partly. The absorption and transport mechanisms of berberine, palmatine, jateorhizine, and coptisine were studied using a Caco-2 cells uptake and transport model, with the addition of cyclosporin A and verapamil as P-glycoprotein (P-gp) inhibitors and MK-571 as a multidrug resistance-associated protein 2 (MRP(2)) inhibitor. In the uptake experiment, berberine, palmatine, jateorhizine, and coptisine were all taken into Caco-2 cells, and their uptakes were increased in the presence of cyclosporin A or verapamil. In the transport experiment, P(app) (AP-BL) was between 0.1 and 1.0 x 10(6) cm/sec for berberine, palmatine, jateorhizine, and coptisine and was lower than P(app) (BL-AB). ER values were all >2. Cyclosporin A and verapamil both increased P(app) (AP-BL) but decreased P(app) (BL-AB) for berberine, palmatine, jateorhizine, and coptisine; ER values were decreased by >50%. MK-571 had no influence on the transmembrane transport of berberine, palmatine, jateorhizine, and coptisine. At a concentration of 1-100 uM, berberine, palmatine, jateorhizine, and coptisine had no significant effects on the bidirection transport of Rho123. Berberine, palmatine, jateorhizine, and coptisine were all P-gp substrates; and at the range of 1-100 uM, berberine, palmatine, jateorhizine, and coptisine had no inhibitory effects on P-gp. Jiao-Tai-Wan (JTW), an important herbal formula consists of Rhizoma coptidis and Cortex cinnamomi powder, is a famous prescription which has been used for centuries to treat insomnia in Traditional Chinese Medicine. The purpose of this study is to compare the pharmacokinetic properties of five protoberberine-type alkaloids (i.e. berberine, palmatine, coptisine, epiberberine and jatrorrhizine), the main bioactive constituents in JTW, between normal and insomnic rats. We also investigate the differences between single-dose and multiple-dose pharmacokinetics of five protoberberine-type alkaloids. The insomnic rat models were induced by intraperitoneal injection of one-dose para-chlorophenylalanine acid (PCPA). Quantification of five protoberberine-type alkaloids in rat plasma was achieved by using a rapid LC-MS/MS method. Plasma samples were collected at different time points to construct pharmacokinetic profiles by plotting drug concentration versus time and estimate pharmacokinetic parameters. An unpaired Student's t test was used for comparisons with SPSS 17.0. The five protoberberine-type alkaloids of single-dose normal groups had slow absorption and low bioavailability, as well as a delay of peak time. In the single-dose oral administration, the Cmax and Tmax of five ingredients in insomnic rats had significant differences compared with those of normal rats. In the multiple-dose oral administration, the pharmacokinetic parameters of five protoberberine-type alkaloids varied greatly in insomnic rats. In the normal rats, there were significant differences (p<0.05) in the principal pharmacokinetic parameters such as Cmax and Tmax between single-dose and multiple-dose oral administration. In the insomnic rats, the five ingredients of multiple-dose groups showed better absorption than the single-dose groups. Particularly, three peaks were observed in multiple-dose model group of plasma-concentration curves. The pharmacokinetic behavior of five protoberberine-type alkaloids was described in this paper. In both normal groups and model groups, the pharmacokinetic behavior of multiple-dose had significant differences comparing with the single-dose; either single-dose or multiple-dose, the pharmacokinetic behavior of insomnic rats had significant differences comparing the normal rats. Multiple dosing may improve the absorption of JTW in insomnic rats, which will increase the bioavailability and bring into active role in therapeutical effect. |
| Toxicity/Toxicokinetics |
- In HFD-fed hamsters, oral administration of Coptisine (50, 100 mg/kg/day) for 4 weeks did not cause significant changes in body weight, food intake, or organ coefficients (liver, kidney, spleen). Serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine (Cr), and urea nitrogen (BUN) were within normal ranges, indicating no obvious hepatotoxicity or nephrotoxicity [2] - Coptisine did not induce obvious cytotoxicity in BV2 cells at concentrations up to 10 μM (detected by MTT assay) [1] |
| References |
[1]. The IDO inhibitor coptisine ameliorates cognitive impairment in a mouse model of Alzheimer's disease. J Alzheimers Dis. 2015;43(1):291-302. [2]. The safety and anti-hypercholesterolemic effect of coptisine in Syrian golden hamsters. Lipids. 2015 Feb;50(2):185-94. [3]. Coptisine-induced cell cycle arrest at G2/M phase and reactive oxygen species-dependent mitochondria-mediated apoptosis in non-small-cell lung cancer A549 cells. Tumour Biol. 2017 Mar;39(3):1010428317694565. |
| Additional Infomation |
Coptisine is an alkaloid. It has a role as a metabolite. Coptisine has been reported in Coptis omeiensis, Corydalis solida, and other organisms with data available. See also: Sanguinaria canadensis root (part of); Chelidonium majus flowering top (part of). - Coptisine is a natural isoquinoline alkaloid derived from Chinese herbal medicines such as Coptis chinensis [1][2][3] - The mechanism of Coptisine ameliorating AD-related cognitive impairment involves inhibiting IDO activity and expression, reducing KYN production, and alleviating Aβ deposition [1] - Coptisine exerts anti-hypercholesterolemic effects by downregulating the expression of hepatic cholesterol synthesis genes (HMGCR, SREBP-2) and lipid accumulation gene (PPARγ) [2] - The anti-tumor effect of Coptisine on A549 cells is mediated by ROS-dependent mitochondria apoptotic pathway and G2/M phase cell cycle arrest [3] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.1219 mL | 15.6094 mL | 31.2188 mL | |
| 5 mM | 0.6244 mL | 3.1219 mL | 6.2438 mL | |
| 10 mM | 0.3122 mL | 1.5609 mL | 3.1219 mL |