Physicochemical Properties
| Molecular Formula | C19H13F4N3O2 |
| Molecular Weight | 391.319038152695 |
| Exact Mass | 391.094 |
| CAS # | 1629729-98-1 |
| PubChem CID | 81689842 |
| Appearance | White to off-white solid powder |
| LogP | 3.3 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 8 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 28 |
| Complexity | 518 |
| Defined Atom Stereocenter Count | 0 |
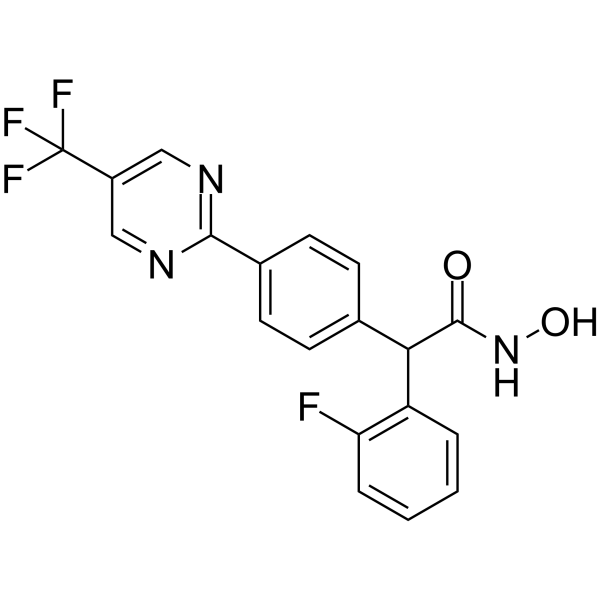
| SMILES | FC1C=CC=CC=1C(C(NO)=O)C1C=CC(C2N=CC(C(F)(F)F)=CN=2)=CC=1 |
| InChi Key | LMGDHGQJJLEAPQ-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C19H13F4N3O2/c20-15-4-2-1-3-14(15)16(18(27)26-28)11-5-7-12(8-6-11)17-24-9-13(10-25-17)19(21,22)23/h1-10,16,28H,(H,26,27) |
| Chemical Name | 2-(2-fluorophenyl)-N-hydroxy-2-[4-[5-(trifluoromethyl)pyrimidin-2-yl]phenyl]acetamide |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | CHDI-390576 has an affinity (Kd) of 80 nM for the immobilised HDAC4 catalytic domain [1]. CHDI-390576 inhibits class I HDAC (1, 3, 8) and class IIb HDAC6 subtypes at IC50s of 39.7 μM, 25.8 μM, 9.1 μM, and 6.2 μM, respectively [1]. |
| ln Vitro |
CHDI-390576 has an affinity (Kd) of 80 nM for the immobilised HDAC4 catalytic domain [1]. CHDI-390576 inhibits class I HDAC (1, 3, 8) and class IIb HDAC6 subtypes at IC50s of 39.7 μM, 25.8 μM, 9.1 μM, and 6.2 μM, respectively [1]. CHDI-390576 inhibited Class IIa HDAC activity in a cellular assay using Jurkat cells with a Boc-Lys(TFA) substrate, showing an IC₅₀ of 69 nM. It showed no observable activity (IC₅₀ > 50 µM) against Class I/IIb HDACs in a parallel cellular assay using Boc-Lys(Ac) as the exogenous substrate. [1] In A549 cells, CHDI-390576 did not show activity against the canonical Class I substrate histone H4K12 acetylation (IC₅₀ > 30 µM) and showed weak activity against the Class IIb substrate tubulin acetylation with an IC₅₀ of 17 µM. [1] The binding affinity (Kd) and kinetics to the catalytic domain of HDAC4 were determined by surface plasmon resonance (SPR), with the Kd value in good agreement with the biochemical IC₅₀. The dissociation half-life of binding to HDAC4 was approximately 3 minutes. [1] At a test concentration of 10 µM, CHDI-390576 exhibited a clean off-target profile, showing <25% displacement of selective radioligands against an extensive panel of CNS and peripheral receptors, ion channels, enzymes, and transporters. [1] CHDI-390576 was stable in mouse and human plasma and blood, and in simulated gastric fluid. [1] |
| ln Vivo |
In a tolerability study in R6/2 transgenic mice (a model of Huntington's disease) and their wild-type littermates, oral administration of CHDI-390576 (10, 30, or 100 mg/kg, b.i.d. for 15 days) was well-tolerated with no significant effects on body weight, body temperature, or observational parameters. [1] Behavioral effects were noted in the open field test. Compared to vehicle control, CHDI-390576 at 30 and 100 mg/kg caused a significant increase in total distance traveled, center distance, and rearing events in both wild-type and R6/2 mice. Average velocity was unchanged. These effects are tentatively attributed to Class IIa HDAC inhibition. [1] |
| Enzyme Assay |
For biochemical HDAC inhibition assays, the catalytic domains of recombinant human HDACs were used. Enzyme activity was measured using fluorogenic peptide substrates: Boc-Lys(TFA)-AMC for Class IIa HDACs (HDAC4, 5, 7, 9) and HDAC8; Ac-Arg-Gly-Lys(Ac)-AMC for HDAC1/2; and Boc-Lys(Ac)-AMC for HDAC3 and HDAC6. Compounds were serially diluted in DMSO and incubated with enzyme and substrate. After a defined period, the reaction was stopped with a developer solution containing trypsin and a deacetylation detection reagent. Fluorescence was measured, and IC₅₀ values were determined from dose-response curves. [1] Surface plasmon resonance (SPR) was used to determine binding kinetics. The catalytic domain of human HDAC4 was immobilized on a sensor chip. Solutions of the compound at various concentrations were flowed over the chip to measure association and dissociation rates, from which the equilibrium dissociation constant (Kd) and half-life were calculated. [1] |
| Cell Assay |
Cellular Class IIa HDAC inhibition was assessed in Jurkat T cells. Cells were treated with serially diluted compounds for a specified duration. Cellular HDAC activity was measured by adding a cell-permeable, fluorogenic substrate, Boc-Lys(TFA)-AMC. After incubation, fluorescence was quantified to determine the IC₅₀ for intracellular Class IIa HDAC inhibition. [1] A counter-screen for Class I/IIb HDAC activity was performed in parallel using the substrate Boc-Lys(Ac)-AMC, which is selective for these classes over Class IIa HDACs in the cellular context. [1] Activity against endogenous HDAC substrates was evaluated in A549 cells. Cells were treated with compound, lysed, and protein extracts were analyzed by western blot using antibodies specific for acetylated histone H4 (Lys12) to assess Class I HDAC inhibition, and for acetylated α-tubulin to assess HDAC6 (Class IIb) inhibition. Band intensity was quantified to determine IC₅₀ values. [1] |
| Animal Protocol |
For pharmacokinetic (PK) studies, fed male C57BL/6 mice were administered CHDI-390576 via intravenous injection (5 mg/kg) or oral gavage (10, 30, or 100 mg/kg). For oral administration, the compound was formulated in a suitable vehicle (specific vehicle not detailed in the main text). Blood (for plasma), brain, and muscle tissues were collected at various time points post-dose for bioanalysis. [1] For the tolerability and behavioral study in R6/2 mice, transgenic mice and wild-type littermates were dosed orally twice daily (b.i.d.) with CHDI-390576 at 10, 30, or 100 mg/kg, or with vehicle, for 15 days. Body weight and temperature were monitored. Open field activity was assessed to measure locomotor and exploratory behavior. [1] |
| ADME/Pharmacokinetics |
Following a single 10 mg/kg oral dose in mice, CHDI-390576 showed total plasma exposure (AUC) of 1960 nM·h and total brain exposure (AUC) of 293 nM·h. It demonstrated oral bioavailability of 44%. [1] The volume of distribution (Vss) was 7.3 L/kg, indicating extensive tissue distribution. Plasma clearance (CL) was 3.1 L/h/kg. [1] The unbound fraction in plasma (fu,plasma) was 4.1%, and the unbound fraction in brain homogenate (fu,brain) was 0.5%. The unbound brain-to-plasma partition ratio (Kp,uu,brain) at Cmax was 0.14. [1] In oral dose escalation studies (10, 30, 100 mg/kg), increases in plasma and brain exposure were approximately dose-proportional. At doses ≥30 mg/kg, total exposure in plasma and brain exceeded the cellular Class IIa HDAC IC₅₀ for approximately 8 hours. [1] The estimated unbound brain Cmax following a 100 mg/kg oral dose was equivalent to the cellular Class IIa IC₃₅ (35 nM). [1] The compound showed high intrinsic clearance in mouse hepatocytes (383 mL/min/kg). [1] Distribution was approximately equal between blood and plasma. [1] |
| Toxicity/Toxicokinetics |
CHDI-390576 showed no significant inhibition (IC₅₀ > 10 µM, max inhibition 30%) of the hERG potassium channel in an IonWorks assay. [1] It showed no inhibition against major human CYP450 isoforms (1A2, 2C8, 2C9, 2C19, 2D6, 3A4) at 50 µM. [1] The compound tested negative in the Ames test, indicating no potential genetic toxicity. [1] In a 15-day repeated-dose study in mice, CHDI-390576 was well-tolerated at doses up to 100 mg/kg b.i.d. with no adverse effects on body weight, body temperature, or general health observations. [1] |
| References |
[1]. Development and characterization of a CNS-penetrant benzhydryl hydroxamic acid class IIa histone deacetylase inhibitor. Bioorg Med Chem Lett. 2019 Jan 1;29(1):83-88. |
| Additional Infomation |
CHDI-390576 is a CNS-penetrant benzhydryl hydroxamic acid inhibitor developed to selectively occupy the catalytic site of Class IIa HDACs (HDAC4, 5, 7, 9). [1] The compound was designed to have improved physicochemical properties (lower topological polar surface area, reduced hydrogen bond donors) to minimize P-glycoprotein (P-gp) efflux and enhance brain exposure, as evidenced by its low effective efflux ratio (EER of 1.2) in MDCK-MDR1 cells. [1] It is presented as a tool compound to evaluate the role of Class IIa HDAC catalytic site occupancy in vitro and in vivo, and to explore its therapeutic potential in neurodegenerative diseases (like Huntington's disease and ALS), neuromuscular, cardiac, and oncology indications. [1] A co-crystal structure of a close analog (compound 16) with the HDAC4 catalytic domain shows the hydroxamic acid chelating the active site zinc ion, and key interactions including a pi-stacking interaction between the pyrimidine capping group and Phe871 of HDAC4. [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~250 mg/mL (~638.86 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (5.32 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (5.32 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (5.32 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5555 mL | 12.7773 mL | 25.5545 mL | |
| 5 mM | 0.5111 mL | 2.5555 mL | 5.1109 mL | |
| 10 mM | 0.2555 mL | 1.2777 mL | 2.5555 mL |