Brilanestrant (formerly known as GDC-0810, ARN-810 and/or RG6046) is a novel, potent and orally bioavailable Selective Estrogen Receptor Degrader (SERD) that is presently being studied in clinical trials for women with locally advanced or metastatic estrogen receptor-positive breast cancer.It has shown strong activity in models of tamoxifen-sensitive and tamoxifen-resistant breast cancer.xenografts of breast cancer resistant to tamoxifen. In contrast to tamoxifen and other ER therapies, GDC-0810 induces a different ERα conformation and does not cause ER agonism in MCF7 cells similar to that of tamoxifen.
Physicochemical Properties
| Molecular Formula | C26H20CLFN2O2 | |
| Molecular Weight | 446.9006 | |
| Exact Mass | 446.12 | |
| Elemental Analysis | C, 69.88; H, 4.51; Cl, 7.93; F, 4.25; N, 6.27; O, 7.16 | |
| CAS # | 1365888-06-7 | |
| Related CAS # |
|
|
| PubChem CID | 56941241 | |
| Appearance | White to off-white solid powder | |
| Density | 1.3±0.1 g/cm3 | |
| Boiling Point | 622.8±55.0 °C at 760 mmHg | |
| Flash Point | 330.4±31.5 °C | |
| Vapour Pressure | 0.0±1.9 mmHg at 25°C | |
| Index of Refraction | 1.690 | |
| LogP | 7.82 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 4 | |
| Rotatable Bond Count | 6 | |
| Heavy Atom Count | 32 | |
| Complexity | 719 | |
| Defined Atom Stereocenter Count | 0 | |
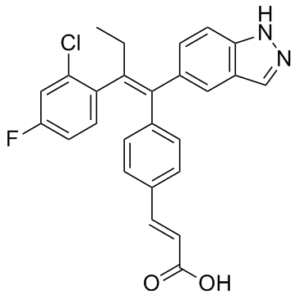
| SMILES | CC/C(=C(/C1=CC=C(C=C1)/C=C/C(=O)O)\C2=CC3=C(C=C2)NN=C3)/C4=C(C=C(C=C4)F)Cl |
|
| InChi Key | BURHGPHDEVGCEZ-KJGLQBJMSA-N | |
| InChi Code | InChI=1S/C26H20ClFN2O2/c1-2-21(22-10-9-20(28)14-23(22)27)26(18-8-11-24-19(13-18)15-29-30-24)17-6-3-16(4-7-17)5-12-25(31)32/h3-15H,2H2,1H3,(H,29,30)(H,31,32)/b12-5+,26-21+ | |
| Chemical Name | (E)-3-[4-[(E)-2-(2-chloro-4-fluorophenyl)-1-(1H-indazol-5-yl)but-1-enyl]phenyl]prop-2-enoic acid | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
estrogen receptor (IC50 = 0.7 nM)
The therapeutic target is estrogen receptor alpha (ER-α/ERα), including wild-type ERα (ER.WT) and mutant ERα (ER.Y537S, ER.D538G); in the FRET-based E2 competitive binding assay, the IC₅₀ values of Brilanestrant for the ligand-binding domains of ER.WT, ER.Y537S, and ER.D538G were detected (specific values not mentioned); in the FRET-based PGC1α recruitment assay, the IC₅₀ values of Brilanestrant for the three ERα subtypes were also detected (specific values not mentioned)[2] The therapeutic target is estrogen receptor alpha (ER-α), with no IC₅₀, Ki, EC₅₀ values mentioned[1] |
| ln Vitro |
GDC-0810 demonstrates a low nanomolar affinity for binding both ERα and ERβ in cell-free radio-ligand competitive binding assays [2]. GDC-0810 exhibits little to no inhibition (IC50 > 20 μM) against CYP1A2, CYP2D6, or CYP3A4. It also has a modest inhibitory effect (IC50 = 2.2 and 3.3 μM, respectively) on CYP2C9 and CYP2C19, and a strong inhibition (IC50 of <0.1 μM) on CYP2C8. It is also discovered that GDC-0810 exhibits good selectivity over other nuclear hormone receptors. GDC-0810 shows negligible activity (IC50 > 1 μM) in transcriptional reporter assays for the mineralocorticoid (MR), progesterone-A (PR-A), progesterone (PR-B), and glucocorticoid (GR) receptors. GDC-0810 shows little activity toward the GR (IC50 = 0.99 μM) and androgen receptor (AR; IC50 > 4 μM) in binding assays[1]. GDC-0810 requires the 26S proteasome in order to deplete ERα. Both in vitro and in vivo, GDC-0810 opposes mutants of the ERα ligand binding domain. Although GDC-0810 has a slightly higher IC50 (WT: 2.6 nM vs. ER.Y537S: 5.5 nM and ER.D538G: 5.4 nM), it still has the ability to potently displace E2 from the ligand binding domain in cell-free E2 competitive binding assays used to determine the binding of GDC-0810 to ER.WT, ER.Y537S, and ER.D538G ligand binding domains. Although it has a ~five-seven fold lower biochemical potency than wild-type ER, GDC-0810 can compete the PGC1α co-activator peptide off the mutated ligand binding domain, suggesting that GDC-0810 can drive a conformational shift of mutant ER from "active" to "inactive"[2]. ERα degradation and antagonistic activity 1. ERα degradation: Brilanestrant induced proteasome-dependent degradation of ERα. After MCF7 cells were treated with 100 nM Brilanestrant for 2, 4, and 6 hours, the ERα protein level decreased significantly; the addition of 26S proteasome inhibitor MG132 (10 μM) reversed this degradation effect[2] 2. ERα antagonistic activity: In-Cell Western assay showed that the inhibitory potency of Brilanestrant on ERα in MCF7 cells was superior to fulvestrant and 4-hydroxytamoxifen (4OH-tamoxifen); mammalian two-hybrid assay indicated that the ERα conformation induced by Brilanestrant was significantly different from that induced by 4OH-tamoxifen and fulvestrant, suggesting a unique mechanism of action[2] 3. Transcriptional activity inhibition: Brilanestrant inhibited the transcription of ER target genes (e.g., PGR, GREB1) in MCF7 cells, and 6-hour treatment significantly downregulated target gene expression (effective both in the presence and absence of 1 nM E2)[2] ### Cell proliferation inhibition 1. MCF7 wild-type cells: In the absence of exogenous estradiol, 5-day treatment with Brilanestrant inhibited MCF7 cell viability in a concentration-dependent manner (CellTiter-Glo luciferase assay), with activity superior to fulvestrant and 4OH-tamoxifen; in the presence of 0.1 nM estradiol, cell proliferation was still significantly inhibited[2] 2. Tamoxifen-resistant cells: Brilanestrant significantly inhibited the proliferation of tamoxifen-resistant MCF7-TamR1 cells[2] 3. ERα mutant cells: Brilanestrant inhibited the proliferation of MCF7 cells expressing ER.Y537S mutation (regardless of the presence of estradiol); it also had proliferation inhibitory activity on T47D ER.D538G knock-in cells, and could selectively inhibit the growth of ER.D538G mutant cells in cell competition experiments[2] ### Endometrial cell activity Ishikawa endometrial cell assay showed that Brilanestrant exhibited only mild estrogenic activity, with the induction effect on alkaline phosphatase activity far lower than estrogenic drugs[2] |
| ln Vivo |
GDC-0810 has a good bioavailability (40–60%) and a low clearance molecule across species, according to its pharmacokinetic profile. The compound has a low to moderate volume of distribution (Vss = 0.2−2.0 L/kg across species) and is highly bound to plasma proteins (>99.5% across species), as would be expected for a lipophilic carboxylic acid. In xenograft models of breast cancer that are both tamoxifen-sensitive and tamoxifen-resistant, GDC-0810 shows strong activity and good bioavailability across species[1]. In both vitro and in vivo uterine models, GDC-0810 exhibits modest estrogenic activity[2]. Tamoxifen-sensitive breast cancer xenograft models 1. MCF7 model: Athymic mice (Crl:NU-Foxn1ⁿᵘ) implanted with 0.36 mg 17β-estradiol slow-release pellets were administered Brilanestrant (1, 10, 100 mg/kg/day, oral) for 28 days, which inhibited tumor growth in a dose-dependent manner; ER target gene expression in tumors of the 100 mg/kg dose group was significantly downregulated, and ¹⁸F-estradiol PET imaging showed reduced ER binding capacity in tumors (45.2% reduction in SUVR in the 10 mg/kg group, 63.3% reduction in the 100 mg/kg group, p<0.0001)[2] 2. HCI-003 patient-derived xenograft (PDX) model: NOD.CB17-Prkdcˢᶜⁱᵈ/NcrCrl mice implanted with 1 mg 17β-estradiol beeswax pellets were administered Brilanestrant (10, 100 mg/kg/day, oral) for 43 days, resulting in significant inhibition of tumor growth; ERα protein level and ER target gene transcription level in tumors of the 100 mg/kg dose group were significantly reduced[2] 3. ZR-75-1 model: 28-day administration of Brilanestrant (100 mg/kg/day, oral) significantly inhibited ZR-75-1 tumor growth, with better efficacy than tamoxifen[2] ### Tamoxifen-resistant breast cancer xenograft models MCF7-TamR1 model (implanted with 0.18 mg 17β-estradiol slow-release pellets) was administered Brilanestrant (10, 30, 100 mg/kg/day, oral) for 27 days, tumor growth was significantly inhibited; in the 100 mg/kg dose group, ERα protein level decreased, Ki67 positive rate decreased, and ER target gene transcription level was significantly downregulated; while tamoxifen (120 mg/kg/day) had no significant tumor inhibitory effect[2] ### ERα mutant tumor models 1. MCF7 HA-ER.Y537S model: Athymic mice without exogenous estradiol supplementation were administered Brilanestrant (100 mg/kg/day, oral) for 28 days, which significantly inhibited tumor growth, with better efficacy than fulvestrant and tamoxifen[2] 2. WHIM20 PDX model (expressing ER.Y537S): Mice without estradiol supplementation were administered Brilanestrant (100 mg/kg/day, oral) for 35 days, tumor growth was significantly inhibited; ERα and PR protein levels in tumors decreased, Ki67 positive rate decreased, and ER target gene expression was downregulated[2] ### In vivo estrogenic activity After administration of Brilanestrant to juvenile rats, uterine wet weight and endometrial cell height only slightly increased, and ER IHC score had no significant elevation, indicating weak in vivo estrogenic effect[2] ### Other in vivo activities Brilanestrant had no tumor inhibitory activity on ER-negative MDA-MB-231 breast cancer xenograft model, confirming its ERα-dependent action[2] |
| Enzyme Assay |
GDC-0810 is a potent ER-α binder (IC50=6.1 nM), a full transcriptional antagonist with no agonism (3× ERE, IC50=2 nM), and displays good potency and efficacy in ER-α degradation (EC50=0.7 nM) and MCF-7 breast cancer cell viability (IC50=2.5 nM) assays. 1. ERα ligand binding assay (FRET-based): FRET-based E2 competitive binding assay was used to detect the binding ability of Brilanestrant to the ligand-binding domains of ER.WT, ER.Y537S, and ER.D538G at E2 concentration of EC80, and IC₅₀ values were calculated; the experiment was repeated multiple times, and results were expressed as mean ± standard deviation[2] 2. ERα-PGC1α interaction assay (FRET-based): FRET-based PGC1α recruitment assay was used to detect the inhibitory effect of Brilanestrant on the interaction between ER.WT/mutants and PGC1α in the presence of agonist (EC80), and IC₅₀ values were calculated; the experiment was repeated multiple times, and results were expressed as mean ± standard deviation[2] |
| Cell Assay |
MCF-7 cells are cultured in RPMI with 10% FBS and 20 mM HEPES at a concentration of 40000 cells per milliliter. Next, a 384-well plate is filled with 16 μL of the cell suspension, containing 640 cells, and the cells are incubated for an entire night to promote cell adhesion. The next day, each compound is added to the cells in 16 μL at a final concentration ranging from 10 to 0.000005 μM through a 10-point, serial 1:5 dilution. Following five days of compound exposure, the cells are treated with 16 μL of CellTiter-GLo, and the relative luminescence units of each well are ascertained. To obtain a background value, 32 μL of medium without cells is mixed with CellTiter-GLo. The following formula is used to calculate each sample's percent viability: (RLU sample-RLU background ×100=%viability; RLU untreated cells-RLU background). ERα degradation detection (Western Blot) 1. Experimental procedure: MCF7 cells were seeded in culture plates and treated with 100 nM Brilanestrant and 100 nM fulvestrant for 2, 4, and 6 hours, with 10 μM MG132 added to some groups; cells were collected to extract total protein, and ERα protein level was detected by Western Blot; biological replicates were set in the experiment, and results were quantified by gray value of protein bands[2] 2. Mutant cell detection: After ER.WT and ER.Y537S MCF7 cells were treated with Brilanestrant for 24 hours, proteins were extracted to detect ERα, PR, and Cyclin D1 levels; after T47D ER.WT/ER.D538G cells were treated with the drug, ERα expression was also detected by Western Blot[2] ### Cell viability detection (CellTiter-Glo assay) 1. Experimental procedure: MCF7 (wild-type/tamoxifen-resistant/ER.Y537S mutant) and T47D (ER.WT/ER.D538G) cells were seeded in 96-well plates and treated with gradient concentrations of Brilanestrant for 5-7 days (without/with 0.1 nM estradiol); CellTiter-Glo reagent was added to detect luciferase activity, and cell viability percentage was calculated (100% for vehicle control); biological quadruplicates were set in the experiment, and results were expressed as mean ± standard deviation[2] ### Immunofluorescence detection (In-Cell Western) 1. Experimental procedure: MCF7 cells were seeded in 96-well plates and treated with gradient concentrations of Brilanestrant, fulvestrant, and 4OH-tamoxifen for 4 hours; after cell fixation, ERα-specific primary antibody and fluorescent secondary antibody were added, and ERα level was quantified by immunofluorescence; triple replicates were set in the experiment, results were expressed as fluorescence intensity, and error was standard error (SEM)[2] ### Gene expression detection (qRT-PCR) 1. Experimental procedure: Cells/tumor tissues treated with Brilanestrant were collected to extract RNA and reverse-transcribe into cDNA; Taqman probe method/qRT-PCR was used to detect the mRNA expression levels of ER target genes (PGR, GREB1, Cyclin D1, etc.); results were normalized to reference genes, based on vehicle control (set to 1), and analyzed by log2 transformation[2] ### Cell competition experiment 1. Experimental procedure: Fluorescently labeled T47D ER.WT (red) and T47D ER.D538G (green) cells were seeded in culture plates at a ratio of 1:1 and cultured in the presence/absence of estradiol and Brilanestrant; red/green fluorescent cell numbers were quantified by regular imaging to evaluate the selective inhibitory effect of the drug on mutant cells; 4 images were taken for each condition, and results were expressed as cell number ratio[2] |
| Animal Protocol |
Subcutaneous implants of time-release pellets containing 0.72 mg 17-β estradiol are made in nu/nu mice. MCF-7 cells are cultured at 37°C with 5% CO2 in RPMI supplemented with 10% FBS. Trypsinized cells are separated and then resuspended at a density of 1×107 cells/mL in 50% RPMI serum-free and 50% Matrigel. Two to three days after pellet implantation, MCF-7 cells are subcutaneously injected (100 μL/animal) into the right flank. Every two weeks, the tumor volume (length × width/2) is measured. When tumors get to about 200 mm3 on average, treatment is initiated and the animals are randomized. For four weeks, animals are given daily treatments with a vehicle or compound. Throughout the trial, tumor volume and body weight are recorded every two weeks. Breast cancer xenograft model (MCF7 wild-type) 1. Experimental animals: Crl:NU-Foxn1ⁿᵘ athymic mice, ovariectomized and implanted with 0.36 mg/60-day release 17β-estradiol slow-release pellets; 1×10⁷ MCF7 cells were inoculated, and mice were randomly grouped 8 days later[2] 2. Administration regimen: Vehicle group (9% Peg-400:0.5% Tween-80:0.5% Povidone:90% 0.5% carboxymethylcellulose, oral), tamoxifen group (60 mg/kg/day, oral), fulvestrant group (200 mg/kg, 3 times a week, subcutaneous), Brilanestrant group (1, 10, 100 mg/kg/day, oral), continuous administration for 28 days[2] 3. Observation indicators: Tumor volume, body weight; tumor tissues were collected at experimental endpoint to detect ER target gene expression and ERα protein level; ¹⁸F-estradiol PET imaging was performed on some mice (1-2 hours after dosing on day 7 of administration)[2] ### Tamoxifen-resistant model (MCF7-TamR1) 1. Experimental animals: Crl:NU-Foxn1ⁿᵘ athymic mice, implanted with 0.18 mg/60-day release 17β-estradiol slow-release pellets, inoculated with MCF7-TamR1 tumor fragments[2] 2. Administration regimen: Vehicle group, tamoxifen group (120 mg/kg/day, oral), fulvestrant group (200 mg/kg, 3 times a week, subcutaneous), Brilanestrant group (10, 30, 100 mg/kg/day, oral), continuous administration for 27 days[2] 3. Observation indicators: Tumor volume, ERα/Ki67 IHC score, ER target gene expression[2] ### Patient-derived xenograft model (HCI-003) 1. Experimental animals: NOD.CB17-Prkdcˢᶜⁱᵈ/NcrCrl mice, implanted with HCI-003 tumor fragments and 1 mg 17β-estradiol beeswax pellets, ovariectomized[2] 2. Administration regimen: Vehicle group, tamoxifen group (60 mg/kg/day, oral), fulvestrant group (200 mg/kg, 3 times a week, subcutaneous), Brilanestrant group (10, 100 mg/kg/day, oral), continuous administration for 43 days; estradiol pellets were removed from one vehicle group as control[2] 3. Observation indicators: Tumor volume, ERα protein level, ER target gene expression[2] ### ERα mutant PDX model (WHIM20) 1. Experimental animals: Specific pathogen-free (SPF) grade mice, without estradiol supplementation, inoculated with WHIM20 tumor fragments to approximately 200 mm³[2] 2. Administration regimen: Vehicle group, tamoxifen group (60 mg/kg/day, oral), fulvestrant group (50 mg/kg on days 1, 3, 8, then 25 mg/kg once a week, subcutaneous), Brilanestrant group (100 mg/kg/day, oral), continuous administration for 35 days[2] 3. Observation indicators: Tumor volume, ER/PR/Ki67 IHC score, ER target gene expression[2] ### Juvenile rat estrogenic activity experiment 1. Experimental animals: Juvenile female rats[2] 2. Administration regimen: Single dose of different concentrations of Brilanestrant, 17α-estradiol (0.1 mg/kg, positive control), or vehicle; 2 mice per group were set in the experiment[2] 3. Observation indicators: Uterine wet weight, endometrial cell height (digital measurement under 20× microscope, 3 measurements per sample), ER IHC score, uterine tissue gene expression (Fluidigm panel)[2] ### Other animal experiments Reference [1] only mentioned that Brilanestrant was administered orally and exhibited activity in tamoxifen-resistant models, without describing specific experimental procedures[1] |
| ADME/Pharmacokinetics |
1. Oral bioavailability: Brilanestrant is a novel non-steroidal, orally bioavailable SERD, with better pharmacokinetic properties compared to fulvestrant which requires intramuscular injection[1][2] 2. Plasma concentration: In the MCF7 xenograft model, effective drug concentration was detected in mouse plasma after 28-day oral administration of Brilanestrant at 100 mg/kg/day[2] 3. Other pharmacokinetic parameters: No specific parameters such as absorption, distribution, metabolism, excretion, and half-life were mentioned[1][2] |
| Toxicity/Toxicokinetics |
1. In vivo toxicity: In all animal experiments, no obvious systemic toxicity (e.g., weight loss, organ damage) was observed when Brilanestrant was administered at doses up to 100 mg/kg/day (oral); only mild uterine proliferative effect was observed in juvenile rats after administration, without significant inflammation or tissue damage[2] 2. Other toxicity: No information on median lethal dose, hepatorenal toxicity, drug-drug interactions, plasma protein binding rate, etc. was mentioned[1][2] |
| References |
[1]. Identification of GDC-0810 (ARN-810), an Orally Bioavailable Selective Estrogen Receptor Degrader (SERD) that Demonstrates Robust Activity in Tamoxifen-Resistant Breast Cancer Xenografts. J Med Chem. 2015 Jun 25;58(12):4888-904. [2]. The selective estrogen receptor downregulator GDC-0810 is efficacious in diverse models of ER+ breast cancer. Elife. 2016 Jul 13;5. pii: e15828. doi: 10.7554/eLife.15828. |
| Additional Infomation |
ARN-810 has been used in trials studying the basic science and treatment of Breast Cancer. Brilanestrant is an orally available, nonsteroidal selective estrogen receptor degrader (SERD), with potential antineoplastic activity. Upon oral administration, brilanestrant binds to the estrogen receptor and induces a conformational change that results in the degradation of the receptor. This may inhibit the growth and survival of ER-expressing cancer cells. 1. Drug category: Brilanestrant (GDC-0810/ARN-810) is a novel non-steroidal, orally bioavailable selective estrogen receptor downregulator (SERD), developed by prospectively optimizing ERα degradation, antagonistic activity and pharmacokinetic properties[1][2] 2. Mechanism of action: Brilanestrant induces unique conformational changes in ERα, both antagonizing the transcriptional activity of ERα and degrading ERα via the proteasome pathway; its action is independent of estradiol, and it still has potent inhibitory effect on ERα mutants (Y537S, D538G)[2] 3. R&D background: Approximately 80% of breast cancers are ERα-positive, and patients are prone to develop resistance to antihormonal therapies such as tamoxifen and aromatase inhibitors, and resistant tumors often still rely on ERα (e.g., ESR1 mutations); although fulvestrant is a SERD, it requires intramuscular injection, limiting the administered dose, and Brilanestrant was developed to solve the above problems[1] 4. Clinical progress: Brilanestrant has entered Phase II clinical trials for the treatment of locally advanced or metastatic ER-positive breast cancer[1][2] 5. Specificity: Brilanestrant is only effective against ERα-positive breast cancer, with no activity against ER-negative tumors, confirming its target specificity[2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.59 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.59 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 3: 2.08 mg/mL (4.65 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2376 mL | 11.1882 mL | 22.3764 mL | |
| 5 mM | 0.4475 mL | 2.2376 mL | 4.4753 mL | |
| 10 mM | 0.2238 mL | 1.1188 mL | 2.2376 mL |