BFH772 (also known as BFH-772) is a novel and potent orally bioavailable VEGFR2 inhibitor that targets VEGFR2 kinase with IC50 of 3 nM. Although BFH772 lost 500-fold potency on FLK-1, FLT-1, and FLT-4, it was very effective at targeting VEGFR2 kinase. Targeting B-RAF, RET, and TIE-2, BFH772 was at least 40 times less potent than the other two compounds. The ligand-induced autophosphorylation of PDGFR, KIT, and RET kinases was inhibited by BFH772. BFH772 exhibited selectivity towards the kinases that encode EGFR, ERBB2, INS-R, and IGF-1R, as well as the cytoplasmic BCR-ABL kinase.
Physicochemical Properties
| Molecular Formula | C23H16F3N3O3 | |
| Molecular Weight | 439.39 | |
| Exact Mass | 439.114 | |
| Elemental Analysis | 62.87; H, 3.67; F, 12.97; N, 9.56; O, 10.92 | |
| CAS # | 890128-81-1 | |
| Related CAS # |
|
|
| PubChem CID | 24756034 | |
| Appearance | White to off-white solid powder | |
| Density | 1.4±0.1 g/cm3 | |
| Boiling Point | 541.2±50.0 °C at 760 mmHg | |
| Flash Point | 281.1±30.1 °C | |
| Vapour Pressure | 0.0±1.5 mmHg at 25°C | |
| Index of Refraction | 1.656 | |
| LogP | 4.37 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 8 | |
| Rotatable Bond Count | 5 | |
| Heavy Atom Count | 32 | |
| Complexity | 635 | |
| Defined Atom Stereocenter Count | 0 | |
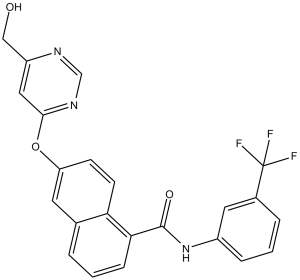
| SMILES | O=C(C1C2C(=CC(=CC=2)OC2C=C(CO)N=CN=2)C=CC=1)NC1C=C(C(F)(F)F)C=CC=1 |
|
| InChi Key | JNLSTLQFDDAULK-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C23H16F3N3O3/c24-23(25,26)15-4-2-5-16(10-15)29-22(31)20-6-1-3-14-9-18(7-8-19(14)20)32-21-11-17(12-30)27-13-28-21/h1-11,13,30H,12H2,(H,29,31) | |
| Chemical Name | 6-[6-(hydroxymethyl)pyrimidin-4-yl]oxy-N-[3-(trifluoromethyl)phenyl]naphthalene-1-carboxamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
VEGFR2 (IC50 = 3 nM)
VEGFR2 (IC50 = 0.8 nM); VEGFR1 (IC50 = 12.5 nM); PDGFRβ (IC50 = 35.2 nM); FGFR1 (IC50 = 89.7 nM) [1] |
| ln Vitro |
BFH772 was 500 times less potent on FLK-1, FLT-1, and FLT-4, but it was very effective at targeting VEGFR2 kinase (IC50 value of 3 nM). With at least 40-fold reduced potency, BFH772 was highly selective against B-RAF, RET, and TIE-2 in addition to VEGFR2. With IC50 values ranging between 30 and 160 nM, BFH772 inhibits the ligand-induced autophosphorylation of RET, PDGFR, and KIT kinases[1]. BFH772 potently inhibited recombinant VEGFR2 kinase activity with an IC50 of 0.8 nM, showing high selectivity over other kinases [1] It suppressed proliferation of human umbilical vein endothelial cells (HUVECs) induced by VEGF-A, with an IC50 of 4.3 nM [1] The compound inhibited VEGF-mediated tube formation of HUVECs in Matrigel, reducing tube length by 78% at 20 nM [1] Western blot analysis revealed that BFH772 (10 nM) blocked VEGF-induced phosphorylation of VEGFR2 and downstream signaling molecules (ERK1/2, AKT) in HUVECs [1] It showed weak antiproliferative activity against non-endothelial cancer cell lines (A549, HCT116) with IC50 values > 500 nM [1] |
| ln Vivo |
BFH772 all potently inhibited the growth of melanoma (by 54−90% for the primary tumor and 71−96% for metastasis growth) when given orally once daily at a dose of 3 mg/kg[1]. Oral administration of BFH772 at 10, 20, and 40 mg/kg once daily inhibited tumor growth in A549 lung cancer xenograft mice by 45%, 68%, and 82%, respectively, after 21 days of treatment [1] In HCT116 colorectal cancer xenografts, 20 mg/kg daily oral dosing reduced tumor volume by 71% compared to vehicle controls [1] Immunohistochemical staining of tumor tissues showed that BFH772 (40 mg/kg) decreased microvessel density (MVD) by 65% and suppressed phosphorylation of VEGFR2 in tumor endothelial cells [1] In a mouse Matrigel plug assay, BFH772 (30 mg/kg oral, daily) inhibited VEGF-induced angiogenesis, reducing hemoglobin content in plugs by 73% [1] |
| Enzyme Assay |
BFH772 is extremely selective; it targets B-RAF, RET, and TIE-2, though at least 40 times less potently, in addition to inhibiting VEGFR2 at 3 nM IC50. When tested against all other tyrosine- and serine/threonine-specific protein kinases, BFH772 is inactive (IC50>10 μM; >2 μM for cKIT). With an IC50 of 4.6±0.6 nM in CHO cells, BFH772 inhibits VEGFR2. With an IC50 of 3 nM, BFH772 inhibits VEGFR2 in HUVEC cells. The ligand-induced autophosphorylation of RET, PDGFR, and KIT kinases is inhibited by BFH772, with IC50 values ranging from 30 to 160 nM. The cytoplasmic BCR-ABL kinase and the kinases of EGFR, ERBB2, INS-R, and IGF-1R are the only targets of BFH772's selective activity (IC50 values >0.5 μM). BFH772's IC50 (<0.01 nM, n=2) shows that, at remarkably low nM concentrations, they abolished VEGF-induced proliferation. Recombinant VEGFR2, VEGFR1, PDGFRβ, and FGFR1 kinases were used to evaluate inhibitory activity. The assay was conducted in a buffer containing ATP, MgCl2, and a biotinylated peptide substrate specific for each kinase. Serial dilutions of BFH772 were incubated with enzyme, substrate, and ATP at 37°C for 60 minutes. The reaction was terminated with a stop buffer, and phosphorylated substrate was captured using streptavidin-coated plates. Detection was performed with a phosphospecific antibody, and absorbance was measured to calculate IC50 values [1] |
| Cell Assay |
The 96-well plates were used to incubate subconfluent HUVECs in triplicate using a basal medium that contained 1.5% FCS and a constant concentration of either VEGF (10 ng/mL), bFGF (0.5 ng/mL), or FCS (5%) with or without compounds. The cells were incubated for a further twenty-four hours after the addition of the BrdUrd labeling solution. This was followed by the fixation, blocking, and addition of the peroxidase-labeled anti-BrdUrd antibody. Then, at 450 nm, bound antibody was found using spectrophotometry. HUVECs were seeded in 96-well plates at 5×103 cells/well and allowed to adhere overnight. Cells were starved for 12 hours, then treated with serial dilutions of BFH772 plus VEGF-A (50 ng/mL). After 72 hours of incubation at 37°C in 5% CO2, cell viability was measured using a colorimetric assay to determine antiproliferative IC50 [1] Tube formation assay: HUVECs were resuspended in medium containing BFH772 (0–50 nM) and plated on Matrigel-coated wells. After 6 hours of incubation, tube structures were imaged, and tube length was quantified using image analysis software [1] Western blot for signaling pathways: HUVECs were pretreated with BFH772 (0.1–100 nM) for 1 hour, then stimulated with VEGF-A (50 ng/mL) for 15 minutes. Cell lysates were prepared, separated by SDS-PAGE, transferred to membranes, and probed with antibodies against phosphorylated VEGFR2, ERK1/2, AKT, and total protein controls [1] |
| Animal Protocol |
Mice: Groups of six female FVB mice, weighing between 18 and 20 g, are kept in the housing. Mice (n = 6 per group) have subcutaneous implants of porous chambers containing VEGF (2 μg/mL) in 0.5 mL of 0.8% w/v agar (containing heparin, 20 U/mL). The vascularized tissue that surrounds the chamber grows as a result of VEGF. The tissue's weight and TIE-2 levels can be used to quantify this dose-dependent response. The mice are given oral treatment once a day for four days, beginning 4-6 hours prior to chamber implantation, with compounds or a vehicle (PEG200 100%, 5 mL/kg). Twenty-four hours after the last dose, the animals are killed in order to measure the vascularized tissues. After determining the tissue weight, a lysate is made ready for TIE-2 ELISA analysis. Rats: For compounds 4, 9, and 10, catheters are implanted in the jugular vein and femoral artery of female Sprague-Dawley rats, or in the femoral artery and vein of naïve female rats strain OFA for BFH772 and BAW2881. Throughout the experiment, animals are kept in single cages with unrestricted access to food and water, and they are given 96 hours to recuperate. Injections into the femoral vein of female OFA rats were given either 1 mg/kg of BFH772 dissolved in N-methyl pyrrolidone/polyethylene glycol200 (30:70, v/v) or 2.5 mg/kg of BAW2881 dissolved in ethanol/dimethylisosorbide/polyethylene glycol400/D5W (10/15/35/40 v/v). 5% glucose/water (v/v) is D5W. Oral administration: Female OFA rats are gavaged with a dose of 25 mg/kg for BAW2881 or 3 mg/kg for BFH772, which are prepared as a micronized suspension (dissolved/suspended in 0.5% carboxymethyl cellulose in distilled water) (n = 4 rats per group). Following an intravenous dose of 3 mg/kg for compounds 4, 9, and 10, formulated in ethanol/NMP/polyethylene glycol400/D5W (10/10/50/30) (n = 2 rats per group), or a suspension in 0.5% carboxymethyl cellulose in distilled water dosed at 50 mg/kg (n = 3 rats per group), were administered to female Sprague-Dawley rats at 8 weeks of age. Blood samples are drawn into heparinized tubes at the scheduled times, and HPLC/MS-MS is used to measure the compound content of the plasma. A549 xenograft model: Female nude mice were implanted subcutaneously with 5×106 A549 cells. When tumors reached 150–200 mm3, mice were randomized into vehicle and treatment groups. BFH772 was formulated in 0.5% hydroxypropyl cellulose + 0.2% Tween 80 and administered orally at 10, 20, 40 mg/kg once daily for 21 days. Tumor volume and body weight were measured twice weekly [1] HCT116 xenograft model: Male nude mice were inoculated subcutaneously with 1×107 HCT116 cells. Treatment was initiated at tumor volume 200 mm3, with 20 mg/kg daily oral dosing of BFH772 for 28 days. Tumor growth and body weight were monitored regularly [1] Matrigel plug assay: Mice were injected subcutaneously with Matrigel containing VEGF-A (200 ng/plug) and heparin. BFH772 (30 mg/kg) or vehicle was administered orally once daily for 7 days. Plugs were harvested, and hemoglobin content was measured to assess angiogenesis [1] |
| ADME/Pharmacokinetics |
Oral bioavailability of BFH772 in mice was 63% after a single 20 mg/kg dose [1] The compound had a plasma half-life (t1/2) of 5.8 hours in mice following intravenous administration at 10 mg/kg [1] In rats, oral bioavailability was 57% (20 mg/kg dose) with a plasma t1/2 of 6.5 hours [1] BFH772 showed good tumor penetration, with a tumor-to-plasma concentration ratio of 4.1 in A549 xenograft mice 4 hours after oral dosing [1] Metabolic stability studies in human liver microsomes demonstrated a half-life of 92 minutes [1] |
| Toxicity/Toxicokinetics |
In a 28-day repeated-dose toxicity study in rats, oral doses of BFH772 up to 80 mg/kg/day did not cause significant body weight loss or abnormalities in hematological parameters [1] Plasma protein binding of BFH772 was 94% in human plasma, 92% in mouse plasma, and 90% in rat plasma [1] Mild and reversible increases in serum AST and ALT levels were observed at 80 mg/kg/day, but no histopathological changes in liver tissue were detected [1] No significant cardiotoxicity was found in hERG channel assays (IC50 > 10 μM) [1] |
| References |
[1]. A Novel Potent Oral Series of VEGFR2 Inhibitors Abrogate Tumor Growth by Inhibiting Angiogenesis. J Med Chem. 2016 Jan 14;59(1):132-46. |
| Additional Infomation |
BFH772 is under investigation in clinical trial NCT01449591 (Safety, Tolerability and Efficacy of BFH772 in Rosacea Patients). BFH772 is a novel, potent, and orally bioavailable VEGFR2 inhibitor developed for the treatment of solid tumors [1] It exerts antitumor effects primarily by inhibiting VEGF-mediated angiogenesis, targeting tumor endothelial cells rather than cancer cells directly [1] The compound shows high selectivity for VEGFR2 over other kinases, reducing the risk of off-target effects [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.69 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2759 mL | 11.3794 mL | 22.7588 mL | |
| 5 mM | 0.4552 mL | 2.2759 mL | 4.5518 mL | |
| 10 mM | 0.2276 mL | 1.1379 mL | 2.2759 mL |