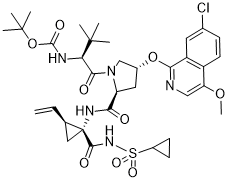
Asunaprevir (formerly BMS-650032; BMS-650032; trade name in Japan and Russia: Sunvepra) is a potent and orally bioavailable small molecule inhibitor of the hepatitis C virus (HCV) NS3 (non-structural) protease that has been approved for use in Japan as part of a combination treatment for HCV infections. It inhibits HCV NS3 protease with an IC50 of 0.2 nM-3.5 nM. HCV serine protease NS3 is required for protein processing during viral replication. Asunaprevir is an experimental drug candidate for the treatment of hepatitis C. It is being tested in combination with pegylated interferon and ribavirin, as well as in interferon-free regimens with other direct-acting antiviral agents including daclatasvir. It is currently in Phase III clinical trials.
Physicochemical Properties
| Molecular Formula | C35H46CLN5O9S | |
| Molecular Weight | 748.29 | |
| Exact Mass | 747.27 | |
| Elemental Analysis | C, 56.18; H, 6.20; Cl, 4.74; N, 9.36; O, 19.24; S, 4.28 | |
| CAS # | 630420-16-5 | |
| Related CAS # |
|
|
| PubChem CID | 16076883 | |
| Appearance | White to off-white solid powder | |
| Density | 1.4±0.1 g/cm3 | |
| Melting Point | 145-155 ºC | |
| Index of Refraction | 1.616 | |
| LogP | 4.02 | |
| Hydrogen Bond Donor Count | 3 | |
| Hydrogen Bond Acceptor Count | 10 | |
| Rotatable Bond Count | 14 | |
| Heavy Atom Count | 51 | |
| Complexity | 1470 | |
| Defined Atom Stereocenter Count | 5 | |
| SMILES | ClC1C([H])=C([H])C2C(=C([H])N=C(C=2C=1[H])O[C@@]1([H])C([H])([H])N(C([C@]([H])(C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])N([H])C(=O)OC(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])=O)[C@@]([H])(C1([H])[H])C(N([H])[C@]1(C(N([H])S(C2([H])C([H])([H])C2([H])[H])(=O)=O)=O)C([H])([H])[C@@]1([H])C([H])=C([H])[H])=O)OC([H])([H])[H] |
|
| InChi Key | XRWSZZJLZRKHHD-WVWIJVSJSA-N | |
| InChi Code | InChI=1S/C35H46ClN5O9S/c1-9-19-16-35(19,31(44)40-51(46,47)22-11-12-22)39-28(42)25-15-21(49-29-24-14-20(36)10-13-23(24)26(48-8)17-37-29)18-41(25)30(43)27(33(2,3)4)38-32(45)50-34(5,6)7/h9-10,13-14,17,19,21-22,25,27H,1,11-12,15-16,18H2,2-8H3,(H,38,45)(H,39,42)(H,40,44)/t19-,21-,25+,27-,35-/m1/s1 | |
| Chemical Name | 1,1-dimethylethyl ((1S)-1-{((2S,4R)-4-(7-chloro-4-methoxyisoquinolin-1-yloxy)-2-({(1R,2S)-1-((cyclopropylsulfonyl)carbamoyl)-2-ethenylcyclopropyl}carbamoyl) pyrrolidin-1-yl)carbonyl}-2,2-dimethylpropyl)carbamate | |
| Synonyms | BMS-650032; BMS 650032; BMS650032; S9X0KRJ00S; 1,1-dimethylethyl ((1S)-1-{((2S,4R)-4-(7-chloro-4-methoxyisoquinolin-1-yloxy)-2-({(1R,2S)-1-((cyclopropylsulfonyl)carbamoyl)-2-ethenylcyclopropyl}carbamoyl) pyrrolidin-1-yl)carbonyl}-2,2-dimethylpropyl)carbamate; trade name in Japan and Russia: Sunvepra | |
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | HCV NS3 protease(IC50≈0.2 nM-3.5 nM) | |
| ln Vitro | Asunaprevir (ASV) inhibits the NS3 proteolytic activity of genotype 1a (H77 strain) and genotype 1b (J4L6S strain), with IC50s of 0.7 and 0.3 nM, respectively. The EC50s of ASV against replicons encoding the NS3 protease domains representing genotypes 1a, 1b, and 4a, range from 1.2 to 4.0 nM. | |
| ln Vivo |
|
|
| Enzyme Assay |
Luciferase assay[1] To monitor IFN signaling directed by ISRE, plasmids pISRE-luc (500 ng/well) expressing firefly luciferase and pRL-TK (50 ng/well) expressing Renilla luciferase as an internal control were co-transfected in Huh 7.5.1 cells (1 × 105). To monitor IFN-β promoter signaling, pGL-4 IFNβ-Luc (pIFNβ/FLuc) (500 ng/well) expressing firefly luciferase and pRL-TK (50 ng/well) expressing Renilla luciferase as an internal control were used (Chang et al., 2006, 2009). Huh 7.5.1 cells were transfected with Lipofectamine 2000 Transfection Reagent following the manufacturer's protocol. Briefly, cells cultured in 12-well plates were transfected with a DNA precipitate containing plasmid DNA (amounts as indicated and had been adjusted by the vector control to be the same among samples of the experimental group). Five hours after transfection, cells were replenished with culture medium, and then incubated for various time points. Relative luciferase activity was assessed by the Promega dual-luciferase reporter assay system. Enzyme-based selectivity assays. [2] To assess in vitro selectivity, compounds were counterscreened against GBV-B NS3/4A protease, human neutrophil elastase, porcine pancreatic elastase, human α-chymotrypsin, human cathepsin A, and human liver cathepsin B. Compounds were diluted in assay buffer in 10% dimethyl sulfoxide (DMSO). GBV-B NS3/4A protease (0.3 nM) replaced HCV-NS3/4A protease in the FRET-based assay, as described above. All other assays were performed in 96-well plates with 1% DMSO and measured continuous substrate hydrolysis at 405 nm on a Spectramax plate spectrophotometer. Human neutrophil elastase, porcine pancreatic elastase, and human pancreatic α-chymotrypsin reaction mixtures contained 50 mM Tris-HCl (pH 8.0), 50 mM NaCl, 0.1 mM EDTA, 0.05% Tween 20, and 20 nM human elastase, 25 nM porcine elastase, or 1 nM chymotrypsin, respectively. Reactions were initiated by addition of substrate (elastases, 5 μM succinyl-AAPV-amino-4-methylcoumarin (AMC); chymotrypsin, 5 μM LLVY-AMC) and monitored fluorimetrically at an excitation wavelength (Ex) of 360 nm and an emission wavelength (Em) of 480 nm. Cathepsin A assays were performed per the manufacturer's instructions with (7-methoxycoumarin-4-yl)acetyl-RPPGFSAFK(dinitrophenyl)-OH as the substrate; reactions were monitored at Ex/Em of 565/580 nm. Cathepsin B assay mixtures contained 50 mM sodium acetate (NaOAc) (pH 5.5), 1 mM tris(2-carboxyethyl)phosphine, 5 nM cathepsin B, and 2 μM Z-FR-AMC as the substrate, and reactions were monitored at Ex/Em of 360/460 nM. Factor XA assay mixtures contained 100 mM NaPO4 (pH 7.5), 0.5% polyethylene glycol (PEG) 8000, 200 mM NaCl, 1 nM human factor XA , and 400 μM S-2222 as the substrate. Factor XIA assay mixtures contained 50 mM HEPES (pH 7.5), 0.1% PEG-8000, 5 mM KCl, 145 mM NaCl, 0.14 nM human factor XIA, and 250 μM S-2366 as the substrate. Factor VIIA assay mixtures contained 50 mM HEPES (pH 7.5), 150 mM NaCl, 5 mM CaCl2, 0.1% PEG-8000, 1.8 nM factor VIIA, and 1 mM S-2288 as the substrate. Kallikrein assay mixtures contained 50 mM Tris (pH 7.5), 100 mM NaCl, 0.5% PEG-8000, 0.2 nM kallikrein (Enzyme Research Laboratories), and 400 μM S-2302 as the substrate. Thrombin assay mixtures contained 100 mM sodium phosphate (pH 7.5), 200 mM NaCl, 0.5% PEG-8000, 0.2 nM thrombin, and 200 μM S-2366 as the substrate. Trypsin assay mixtures contained 100 mM NaPO4 (pH 7.5), 200 mM NaCl, 0.5% PEG-8000, 0.332 nM trypsin, and 200 μM S-2222 as the substrate. Asunaprevir (ASV) inhibits the NS3 proteolytic activity of genotype 1a (H77 strain) and genotype 1b (J4L6S strain), with IC50s of 0.7 and 0.3 nM, respectively. The EC50s of ASV against replicons encoding the NS3 protease domains representing genotypes 1a, 1b, and 4a, range from 1.2 to 4.0 nM. |
|
| Cell Assay |
Cell culture virology assays.[2] For HCV replicons containing an RLuc reporter, compound antiviral activities (50% effective concentration [EC50]), were determined as described previously. For replicons lacking a reporter gene, activity was measured using a FRET-labeled peptide substrate (10 μM), as described previously. BVDV assays were performed as described previously (36), except that incubations were maintained for 4 days. Antiviral susceptibility of HIV, HRV2, and HCoV was determined by incubation with serial dilutions of compound. For recombinant HIVs expressing RLuc, antiviral activity was evaluated by luciferase assay 5 days postinfection using a dual-luciferase kit. The diluted passive lysis solution was premixed with both the luciferase assay substrate and the Stop & Glo substrate (2:1:1 ratio). To each aspirated sample well, 40 μl of the mixture was added, and luciferase activity was measured immediately on a Wallac TriLux. For determination of activities against HRV2 and HCoV OC43, compound and virus (multiplicity of infection, 0.1) were incubated with cells for 96 h, and cell protection was used as a measure of virus production. For the cell protection assay, 17 μl of Cell-Titer Blue reagent was added to each well. Plates were then incubated for 2 h at room temperature before fluorescence was measured using a Spectramax Gemini EM instrument set to Ex/Em of 530/580 nm. Cytotoxicity is determined by incubating cells (3,000 to 10,000 cells/well) with serially diluted test compounds or DMSO for 5 days (MT-2 cells) or 4 days (all other cell types). Cell viability is quantitated using an MTS assay for MT-2 or a Cell-Titer Blue reagent assay for HEK-293, HuH-7, HepG2, and MRC5 cells, and 50% cytotoxic concentrations (CC50s) are calculated. For the HCV and BVDV replicon assays, CC50s are determined from the same wells that are later used to determine EC50s. |
|
| Animal Protocol |
In vivo exposure studies.[2] The amorphous free acid form of Asunaprevir (ASV) was used in all studies. Plasma and tissue exposures were characterized in male FVB mice (20 to 25 g), male Sprague-Dawley rats (300 to 350 g; Hilltop Lab Animals, Scottsdale, PA) with indwelling cannulas implanted in the jugular vein or common bile duct, male beagles (ca. 9 to 12 kg) bearing vascular access ports, and male cynomolgus monkeys bearing vascular access ports (3.1 to 5.7 kg). All animal procedures were performed under protocols approved by the Institutional Animal Care and Use Committee of the test facility. In all studies, coagulated blood samples taken after dosing were centrifuged at 4°C (1,500 to 2,000 × g); plasma samples were stored at −20°C until analyzed. Tissue samples were rinsed, blotted dry, weighed, and stored frozen. ASV in plasma and tissue samples was analyzed by LC-MS/MS. The lower limit of quantification (LLOQ) of ASV was 1 nM in plasma samples from all species, 5 nM in mouse and rat tissues, and 50 nM in dog and monkey tissues.[2] Mice (n = 9 per group; overnight fast) received ASV by oral gavage (5 mg/kg; vehicle of PEG-400–ethanol, 9:1). Blood samples (∼0.2 ml) were obtained by retro-orbital bleeding at 0.25, 0.5, 1, 3, 6, 8, and 24 h after dosing. Within each group, three animals were bled at 0.25, 3, and 24 h, three at 0.5 and 6 h, and three at 1 and 8 h, resulting in a composite pharmacokinetic profile. Livers and brains were also removed from mice at the terminal sampling points.[2] Rats (n = 3 per group; overnight fast) received ASV (amorphous free acid) by oral gavage (3, 5, 10, and 15 mg/kg) in PEG-400–ethanol (9:1). Serial blood samples (∼0.3 ml) were obtained from the jugular vein predosing (0 h) and at 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, 24, and 48 h postdosing. To assess tissue exposure, rats were orally administered ASV (5 or 15 mg/kg, same vehicle as above), and blood, liver, and heart samples from two rats/group were obtained at 0.17, 0.5, 1, 2, 4, 6, 8, 24, 48, and 72 h after dosing.[2] Dogs (n = 3; overnight fast) were administered ASV by oral gavage at 3 or 6 mg/kg (3 mg/ml in 85% PEG-400–15% water). Serial blood samples were collected from vascular access ports at 0.08, 0.167, 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, 24, and 48 h postdose. To assess tissue exposure, six male dogs (8.4 to 12.5 kg) were orally administered ASV (6 mg/kg), and blood, liver, and spleen samples were obtained from one dog each at 1, 3, 7, 24, 48, and 72 h postdosing.[2] Monkeys (n = 3 males; overnight fast) were administered ASV by oral gavage at 3 mg/kg (3 mg/ml in 85% PEG-400–15% water). Blood samples were collected at 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, and 24 h postdose. To assess tissue exposure, three male and six female cynomolgus monkeys (2 to 5 kg) were orally administered ASV (10 mg/kg), and blood, liver, and spleen samples were obtained from one male each at 2, 8, and 24 h postdosing and from one female each at 0.5, 2, 4, 8, 24, and 30 h postdosing.[2] Asunaprevir (ASV) is administered orally to mice (n = 9 per group; overnight fast; vehicle: PEG-400-ethanol, 9:1). Retro-orbital bleeding is used to obtain blood samples (-0.2 mL) at 0.25, 0.5, 1, 3, 6, 8, and 24 hours following dosing. To create a composite pharmacokinetic profile, three animals in each group are bled at 0.25, 3, and 24 hours, three at 0.5 and 6 hours, and three at 1 and 8 hours. At the terminal sampling points, mice are also deprived of their livers and brains.Amorphous free acid (ASV) is given orally to rats (n=3 per group; overnight fast) at doses of 3, 5, 10, and 15 mg/kg in PEG-400-ethanol (9:1). The jugular vein is used to draw serial blood samples (-0.3 mL) prior to dosing (0 h) and at 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, 24, and 48 h after dosing. Rats are given ASV (5 or 15 mg/kg, same vehicle as above) orally in order to measure tissue exposure. Samples of the rats' liver, heart, and blood are taken at intervals of 0.17, 0.5, 1, 2, 4, 6, 8, 24, 48, and 72 hours following dosing. |
|
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion In preclinical studies, asunaprevir showed a high liver-to-plasma AUC ratio. It is rapidly absorbed within 30 minutes of administration. Clinical pharmacokinetic studies showed a Tmax of 2-4 hours. The pharmacokinetic profile act in a dose-proportional manner and in a dose of 100 mg the steady-state Cmax and AUC was 572 ng/ml and 1887 ng x h/mL. The absolute bioavailability is reported to be 9.3%. The absorption of asunaprevir is increased with food. Asunaprevir is primarily eliminated via the feces. From the administered dose, 84% is excreted by feces mainly as metabolites and less than 1% of the dose is recovered as metabolites in the urine. The proportion of unchanged asunaprevir recovered in feces represents only 7.5% of the dose. The registered volume of distribution at steady state is 194 L. Clinical pharmacokinetic studies showed a mean oral clearance of 302-491 L/h. Metabolism / Metabolites Asunaprevir is metabolized by the liver. The metabolism is mainly marked by oxidative reactions mediated by the activity of CYP3A. Asunaprevir seems to weakly induce its own metabolism and from the circulating dose, just about 5% of the administered dose is formed by metabolites. The metabolites of asunaprevir are formed after mono- and bis-oxidation, N-dealkylation, loss of isoquinoline ring and O-demethylation. All the metabolic reactions form about 15 metabolites and studies have reported that the main metabolic activity is performed by CYP3A4 and CYP3A5 with some minor activity from CYP2A6, CYP2B6, CYP2C9, CYP2C19 and CYP2D6. Biological Half-Life Clinical pharmacokinetic studies showed a mean terminal half-life of 15-20 hours. |
|
| Toxicity/Toxicokinetics |
Protein Binding Protein binding of asunaprevir is very high and it can reach more than 99% of the administered dose independently of the dose. _In vitro_ studies with human Caco-2 cells indicated that asunaprevir is a substrate of P-gp, OATP1B1 and OATP2B1. |
|
| References |
[1]. Front Microbiol. 2017; 8: 668 [2]. Antimicrob Agents Chemother.2012 Oct;56(10):5387-96. [3]. Antimicrob Agents Chemother. 2012 Oct;56(10):5230-9. [4]. Antimicrob Agents Chemother. 2012 Oct;56(10):5387-96. [5]. Antimicrob Agents Chemother. 2012 Jul;56(7):3670-81. |
|
| Additional Infomation |
Asunaprevir is an oligopeptide. Asunaprevir, also named BMS-650032, is a potent hepatitis C virus (HCV) NS3 protease inhibitor. It has been shown to have a very high efficacy in dual-combination regimens with daclatasvir in patients chronically infected with HCV genotype 1b. It was developed by Bristol-Myers Squibb Canada and approved by Health Canada on April 22, 2016. The commercialization of asunaprevir was cancelled one year later on October 16, 2017. Asunaprevir is an orally bioavailable inhibitor of the nonstructural protein 3 (NS3), with potential activity against hepatitis C virus (HCV). Upon administration, asunaprevir binds to the active center of the HCV NS3 and prevents NS3 protease-mediated polyprotein maturation. This disrupts the processing of viral proteins required for HCV replication. NS3, a serine protease, is essential for the proteolytic cleavages within the HCV polyprotein and plays a key role during HCV viral RNA replication. HCV is a small, enveloped, single-stranded RNA virus belonging to the Flaviviridae family. Drug Indication Asunaprevir is indicated in combination with other agents for the treatment of chronic hepatitis C in adult patients with hepatitis C virus genotypes 1 or 4 and compensated liver cirrhosis. Hepatitis C is a liver disease caused by the hepatitis C virus. The chronic state of this condition accounts for 60-80% of the cases from which the risk of cirrhosis of the liver within 20 years is of around 15-30%. The genotype 1 is the most common type of hepatitis C in the United States and the most difficult to treat. Treatment of chronic hepatitis C Mechanism of Action Asunaprevir is a highly active HCV NS3 protease inhibitor. The genome of HCV has a positive polarity which allows it to be translated into a protein in the host cell without further transformation steps. However, the resultant protein needs to be divided by the enzyme NS3 protease into single proteins in order to be able to exert its enzymatic activity or structural role. Therefore, due to NS3 vital importance for viral replication, the inhibiting action of asunaprevir causes a robust antiviral activity. Pharmacodynamics Studies in vitro demonstrated a significant antiviral activity in HCV replicon cell systems with an EC50 of 4nm and 1nm against the HCV genotype 1a and 1b respectively. These studies showed a limited activity against the genotypes 2 and 3. This property makes asunaprevir a highly selective anti-HCV agent that is not effective against HCV closely related virus. Asunaprevir produce robust declines in HCV RNA levels in patients with HCV genotype 1 infection.In clinical studies, it has been shown that asunaprevir is well-tolerated and the mean maximum HCV RNA level reduction from baseline was of approximately 2.87 log10 IU/ml. Monotherapy clinical studies with asunaprevir showed a mean maximum decline of HCV RNA in the range of 0.28-2.87 log10 IU/ml when administered in increasing doses from 10-600 mg. When asunaprevir was used as a combination product, it was possible to obtain a sustained virological response (aviremia 24 weeks after completion of therapy) in 83-92% of the patients. |
Solubility Data
| Solubility (In Vitro) |
DMSO : ~100 mg/mL ( ~133.63 mM ) Ethanol : 20~100 mg/mL(~26.73 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (3.34 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (3.34 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (3.34 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 2.5 mg/mL (3.34 mM) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 5: 2 mg/mL (2.67 mM) in 10% EtOH + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.0 mg/mL clear EtOH stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 6: ≥ 2 mg/mL (2.67 mM) (saturation unknown) in 10% EtOH + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.0 mg/mL clear EtOH stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 7: 10% DMSO+40% PEG300+5% Tween-80+45% Saline: ≥ 2.5 mg/mL (3.34 mM) (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.3364 mL | 6.6819 mL | 13.3638 mL | |
| 5 mM | 0.2673 mL | 1.3364 mL | 2.6728 mL | |
| 10 mM | 0.1336 mL | 0.6682 mL | 1.3364 mL |